 2018, Vol. 32
2018, Vol. 32 

舒肝丸由川楝子、醋延胡索、白芍、片姜黄、木香、沉香、豆蔻仁、砂仁、姜厚朴、陈皮、枳壳、茯苓、朱砂共13味中药组成,各药味经粉碎后,加入不同的辅料精制成大蜜丸、小蜜丸、水蜜丸或水丸。临床上主要用于舒肝和胃、理气止痛等。现《中国药典》(2015年版)收载该药品的质量标准[1]中检验项目包括:显微鉴别,朱砂理化鉴别,延胡索、陈皮、厚朴、木香的薄层色谱鉴别及白芍中芍药苷的高效液相色谱法含量测定。该质量标准检验内容在《中国药典》(2005年版)后无相关修订,检验方法较为陈旧。查阅文献报道的质量标准研究内容仅测定芍药苷[2]或延胡索乙素[3]的含量,无定性项检验;参考骨痹止痛液等质量标准完善提高的文献[4-8]中报道的对厚朴、延胡索定性鉴别研究及益胃颗粒等文献[7-11]报道的对芍药苷定量测定的研究,对本品的质量标准进行全面提高完善,修订了延胡索、陈皮、厚朴的薄层色谱鉴别,建立了更高效快捷的芍药苷HPLC法含量测定项。新方法缩短了分析时间、节约了检验成本,提高检验效率,为舒肝丸的质量标准制定提供科学依据。
1 仪器与试药 1.1 仪器Linomat-5半自动点样仪(CAMAG公司);LC-20AT高效液相色谱仪(日本岛津公司);KQ-400KDE超声波清洗仪(昆山市超声仪器有限公司);AE240型电子天平(上海梅特勒仪器有限公司);薄层板:硅胶G薄层板(10×10cm,青岛海洋化工厂分厂);色谱柱:Thermo Acclaim C18(250 mm×4.6 mm,5μm;美国戴安公司)。
1.2 试药延胡索对照药材(批号:120928-201208)、芍药苷对照品(批号:110736-201035,含量96.4%)、橙皮苷对照品(批号:110721-201014,含量95.3%)、厚朴酚对照品(批号:110729-200412)、厚朴酚对照品(批号:110730-201313)均购自中国食品药品检定研究院。
舒肝丸样品三批购自市场(北京同仁堂股份有限公司同仁堂制药厂,批号3015234;哈药集团世一堂制药厂,批号1506107;颈复康药业集团有限公司,批号395034,编号Ⅰ~Ⅲ)。川楝子、醋延胡索、白芍(酒炒)、片姜黄、木香、沉香、豆蔻仁、砂仁、姜厚朴、陈皮、枳壳(炒)、茯苓、朱砂经本院中药室段吉平主任药师鉴定。
甲醇、乙腈(色谱纯,Merck公司);水用去离子水;其它试剂均为分析纯。
2 方法与结果 2.1 薄层色谱鉴别 2.1.1 陈皮取本品水丸2.5 g或水蜜丸4 g,研碎;或取小蜜丸、大蜜丸6 g,剪碎,加甲醇40 mL,加热回流30 min,滤过,取滤液1 mL,作为供试品溶液。另取橙皮苷对照品,加甲醇制成饱和溶液,作为对照品溶液。按处方比例称取除陈皮外其他药材适量,制成缺陈皮的阴性样品,同供试品溶液制备方法制成缺陈皮的阴性对照溶液。照薄层色谱法(附录Ⅵ B)试验,吸取对照品溶液2μL、供试品溶液及阴性对照溶液各5μL,分别点于同一聚酰胺薄膜上,以三氯甲烷-丙酮-甲醇(5:1:1)为展开剂,展开,取出,晾干,喷以三氯化铝试液,置紫外光灯(365 nm)下检视。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点。
新旧方法结果对比见图 1,经验证,所得色谱斑点清晰、圆整、分离较好,且阴性无干扰。

|
图 1 两方法陈皮薄层色谱法鉴别结果 A.为新标准方法;B.为现行标准方法 1.橙皮苷对照;2.陈皮阴性样品;3~5.舒肝丸样品 |
取陈皮鉴别节下剩余滤液蒸干,残渣加水20 mL使溶解,分取10 mL(剩余水液备用),加浓氨试液调制碱性,用乙醚振摇提取2次,每次15 mL,合并乙醚液,挥干,残渣加甲醇1 mL使溶解,作为供试品溶液。另取延胡索对照药材1 g,加甲醇40mL,加热回流30 min,滤过,滤液蒸干,同法制成对照药材溶液。按处方比例称取除延胡索外其他药材适量,制成缺延胡索的阴性样品,同供试品溶液制备方法制成缺延胡索的阴性对照溶液。照薄层色谱法(附录Ⅵ B)试验,吸取上述对照药材溶液5μL、供试品溶液及阴性对照溶液各10μL,分别点于同一硅胶G薄层板上,以环己烷-三氯甲烷-甲醇(7.5:4:1)为展开剂,展开,取出,晾干,置碘蒸气熏至斑点显色清晰,取出,置紫外光灯(365 nm)下检视。供试品色谱中,在与对照药材色谱色谱相应的位置上,显相同颜色的荧光斑点。
新旧方法结果对比见图 2,经验证,所得色谱斑点清晰、圆整、分离较好,且阴性无干扰。

|
图 2 两方法延胡索薄层色谱法鉴别结果 A.为新标准方法;B.为现行标准方法 1.延胡索对照药材;2.延胡索阴性样品;3~5.舒肝丸样品 |
取延胡索鉴别剩余水溶液,加盐酸调至酸性,用乙醚振摇提取2次,每次15 mL,合并乙醚液,挥干,残渣加甲醇1 mL使溶解,作为供试品溶液。取厚朴酚对照品与和厚朴酚对照品,加甲醇制成每1 mL各含1 mg的混合溶液,作为对照品溶液。按处方比例称取除厚朴外其他药材适量,制成缺厚朴的阴性样品,同供试品溶液制备方法制成缺厚朴的阴性对照溶液。照薄层色谱法(附录Ⅵ B)试验,吸取上述对照品溶液5μL、供试品溶液及阴性对照溶液各10μL,分别点于同一硅胶GF254薄层板上,以环己烷-乙酸乙酯(3:1)为展开剂,展开,取出,晾干,置紫外光灯(254 nm)下检视。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。
新旧方法结果对比见图 3,经验证,所得色谱斑点清晰、圆整、分离较好,且阴性无干扰。

|
图 3 两方法厚朴薄层色谱法鉴别结果 A.为新标准方法;B1.为现标准方法(254nm);B2.为现行标准方法(日光) 1.混合对照品;2.阴性样品;3~5.舒肝丸样品 |
色谱柱:Thermo Acclaim C18(250mm× 4.6mm,5μm);流动相:乙腈(A)-0.05%磷酸(B),梯度洗脱(0~40 min,15%A→25%A);柱温:3 5 ℃;检测波长为2 3 0 n m;流速1. 0 ml·min-1;进样量:5μL;理论板数大于8000;芍药苷与相邻峰的分离度均大于1.5。
2.2.2 对照品溶液的制备精密称取芍药苷对照品0.01011 g,置25 mL量瓶中,加甲醇适量使溶解并稀释至刻度,摇匀,精密量取5 mL,置50 mL量瓶中,加甲醇稀释至刻度,摇匀,即得对照品溶液。
2.2.3 供试品溶液的制备取重量差异节下的本品,剪碎,取约0.8g,精密称定,置具塞锥形瓶中,精密加入甲醇25 mL,密塞,称定重量,加热回流90 min,取出,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,即得。
2.2.4 阴性样品溶液的制备按质量标准处方比例及制备工艺,将药材粉碎后,自行配制缺白芍的阴性样品,并按“2.2.3”节下方法制备溶液,即得。
2.2.5 专属性考察分别取对照品溶液、供试品溶液及阴性缺味药材溶液进样分析,按“2.2.1”节下色谱条件进样分析,结果样品色谱中与对照品色谱相同保留时间处有色谱峰,而阴性样品相应位置无相应峰,不干扰测定。待测成分与杂质分离度良好。

|
图 4 芍药苷色谱图 A.对照品;B.样品;C.白芍;D.阴性样品 |
取系列浓度对照品溶液,按拟定的色谱条件测定,记录峰面积。以对照品进样量(μg)为横坐标,峰面积积分值为纵坐标,进行线性回归。结果线性方程为y=1.269×106x+1727.45;线性范围为0.03898~0.9476μg;相关系数为0.9999,表明待测成分线性关系良好。
2.2.7 重复性试验取同一舒肝丸样品约0.64 g、0.8 g、0.96 g各三份,精密称定,按“2.2.3”节下制备9份供试品溶液,分别测定含量,结果样品中芍药苷含量为0.8945 mg·g-1,RSD为1.4%,结果表明方法重复性良好。
2.2.8 稳定性试验取同一供试品溶液,分别在0、2、7、13和20h进样测定,记录峰面积。结果RSD为1.1%,表明供试品溶液中待测成分在室温放置20 h内基本稳定。
2.2.9 回收率试验采用加样回收法,取“2.2.7”项下已知含量的样品0.4 g,共9份,精密称定,分别置具塞锥形瓶中,分别精密加入不同浓度的对照品溶液各25ml,按“2.2.3”节下方法制备供试品溶液,按“2.2.1”节下色谱条件进行分析,计算回收率。结果芍药苷的平均回收率(n=9)为100.5%,RSD为1.5%,表明方法回收率良好,方法可行。
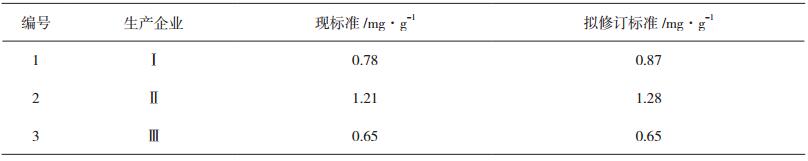
2.2.10 样品测定结果对比取3批舒肝丸样品,分别按现标准[1]及拟修订标准(即按“2.2.3”节下方法制备供试品溶液,按“2.2.1”节下色谱条件进行测定)测定芍药苷含量,结果对比见表 1。
|
|
表 1 样品测定结果对比(n=2) |
三个薄层色谱鉴别均为舒肝丸现行标准收录的检验项目,但方法繁琐,毒性试剂使用率高、使用量大,如陈皮使用甲苯作为展开剂;延胡索的样品前处理用苯加热回流时间为2 h,展开剂系统也使用了苯,费时,不环保,而且按标准检验结果显示薄层色谱斑点不清晰;厚朴样品前处理过程萃取次数达9次,而且频繁使用三氯甲烷,展开系统中也使用了苯。优化后的薄层色谱鉴别方法可以共用1份样品,经过不同极性的溶剂振摇提取后,按样品中化学成分的极性大小富集,分别进行薄层色谱鉴别,简化了操作,大大节约样品用量,大幅节约检验成本。
3.2 含量测定方法的优化现行标准[1]及文献报道[12]的芍药苷高效液相色谱法含量测定中,流动相使用的磷酸缓冲盐(取5份0.067 mol·L-1的磷酸氢二钠溶液,用约1份0.067 mol·L-1的磷酸二氢钾溶液调节pH为7.4)配制繁琐;供试品溶液制备的过程中,样品经过超声提取后,采用大孔树脂柱净化、浓缩,试验过程繁琐。而优化后的含量测定方法,以乙腈-水-磷酸为流动相,样品仅需超声处理,大大提高了工作效率。
3.3 小结舒肝丸现行标准收载项目虽较为完善,但鉴别及含量测定试验中,样品的处理及检测方法操作复杂,薄层色谱鉴别样品提取及薄层板展开过程中苯、甲苯等毒性试剂使用率高,含量测定项中结果易受样品净化过程中柱填料(GDX-201型大孔吸附树脂)影响而致重现性差。因此,探索方便、快捷及更高效的检测方法十分必要,本研究建立了环保高效的薄层色谱鉴别方法及操作简单、重现性好的含量测定方法,为舒肝丸现行标准的提升打下良好基础。
| [1] |
中国药典: 一部[S]. 2015: 1600.
|
| [2] |
孙大鹏. 高效液相色谱法测定舒肝丸中芍药苷含量[J]. 现代中药研究与实践, 2006, 20(2): 32-33. |
| [3] |
朱晓军, 陈彬. 高效液相色谱法测定舒肝丸中延胡索乙素的含量[J]. 医药导报, 2006, 25(6): 570-572. |
| [4] |
李喜香, 李兴勇, 包强, 王雪梅. 骨痹止痛液的质量标准提高研究[J]. 中国药房, 2017(9): 1249-1253. DOI:10.6039/j.issn.1001-0408.2017.09.27 |
| [5] |
丁曼, 华洁, 周福军, 侯文彬. 和胃颗粒质量标准研究[J]. 西北药学杂志, 2016(1): 18-22. |
| [6] |
幸而, 杨银凤, 曾源泉, 陈水仙. 舒肝健胃丸的质量标准研究[J]. 今日药学, 2016(11): 783-785. |
| [7] |
程艳芹, 刘旭, 赵丽艳, 李明春. 益胃颗粒的质量标准研究[J]. 中国药房, 2015(36): 5123-5125. |
| [8] |
刘冰, 祝小静. 舒肝调气丸质量标准研究[J]. 天津药学, 2010, 22(6): 15-18. |
| [9] |
毛和平, 刘光斌, 石晓峰, 等. 柴胡舒肝健乳丸质量标准研究[J]. 西部中药, 2013, 26(10): 31-33. |
| [10] |
贾福群, 刘卫东, 任建国. 通脉颗粒的质量标准研究[J]. 西北药学杂志, 2017(2): 130-133. |
| [11] |
王芳, 刘皈阳, 马建丽, 等. HPLC法测定丹膝颗粒中芍药苷的含量[J]. 中国药事, 2013, 27(2): 199-201. |
| [12] |
杜春双, 李宏, 宋晓坤. HPLC法测定乳痛丸中芍药苷的含量[J]. 中国药事, 2012, 26(4): 385-387. |