 2018, Vol. 32
2018, Vol. 32 

2. 广西壮族自治区食品药品检验所, 南宁 530021
2. Guangxi Institute for Food and Drug Control, Nanning 530021, China
茶苯海明为抗组胺药,为苯海拉明(1, 3-二甲基-8-氯-1H-嘌呤-2, 6-二酮)和8-氯茶碱[N, N-二甲基-2-(二苯基甲氧基)乙胺]1︰1的混合物(见图 1),收载于《中国药典》2015年版二部[1],规定本品按干燥品计,含苯海拉明应为53.0%~55.5%,含8-氯茶碱应为44.0%~47.0%。此外,美国药典38版[2]、欧洲药典8.0版[3]及日本药典17版[4]都收载该品种,其含量测定方法均为分别采用高氯酸滴定液滴定苯海拉明的含量、采用硫氰酸铵滴定液滴定8-氯茶碱的含量,方法较为繁琐且专属性不强。

|
图 1 茶苯海明化学结构 A.苯海拉明;B. 8-氯茶碱。 |
在对照品标化中,为保证赋值准确,通常采用多种含量测定方法如HPLC外标法、滴定法、紫外光谱法等测定结果来相互佐证。在标化茶苯海明对照品时,经查阅文献[5]报道了HPLC-UV梯度洗脱的方法对茶苯海明及其杂质进行测定,考虑到苯海拉明和8-氯茶碱的紫外响应可能不同,故需要建立新的方法来测定其含量。近年来,核磁定量法(qNMR法)技术日趋成熟,并因其具有绝对定量的优势[6-7],被越来越多地应用于标准物质的含量测定中,甚至混合物的测定中[8-14],因此,本研究建立了qNMR法,首次同时测定了茶苯海明中苯海拉明与8-氯茶碱两组分的绝对含量,方法快速简单、准确且专属性强。
1 仪器与试药 1.1 仪器Avance DRX 500型核磁共振谱仪(瑞士布鲁克公司),Mettler Toledo XP230分析天平。
1.2 试药氘代甲醇(J & K Scientific, 批号:182473);对苯二酚(ACROS公司,纯度99.5%)。茶苯海明原料药(上海万代制药有限公司,批号:1507014);茶苯海明对照品(中国食品药品检定研究院,批号:100120-200403,含量:99.7%)。高氯酸滴定液(0.1 mol·L-1)、硝酸银滴定液(0.1 mol·L-1)、硫氰酸铵滴定液(0.1 mol·L-1)均为实验室自制。
2 方法与结果 2.1 核磁定量方法 2.1.1 样品制备精密称取一定量的内标(对苯二酚)及茶苯海明适量,加入一定体积的氘代甲醇配成浓度均为0.05 mol·L-1的溶液,作为供试品溶液;同法制备对照品溶液;分别转入5 mm核磁管中备用。
2.1.2 核磁共振定量核磁共振(NMR)采集条件:采用zg30脉冲序列在恒温(25 ℃)下获取1H NMR谱。具体试验参数:谱宽(SWH)10000 Hz,采样点数(TD)64K,采样时间(AQ)3.28 s,驰豫延迟时间(D1)20 s,采样次数(NS)16次,空扫次数(DS)2次,采用手动积分,每个峰积分3次,相对标准偏差(RSD)<1%时取平均值,测定样品和内标选定峰峰面积的相对比值,采用《中国药典》 2015年版附录0441中绝对定量公式进行计算[15]。
2.1.3 氘代溶剂与内标物的选择根据样品的溶解性选择了氘代甲醇作为溶剂,且溶剂峰与茶苯海明中的苯海拉明与8-氯茶碱的定量峰互不干扰。
根据茶苯海明的1H-NMR的归属以及CD3OD的溶解性,选择了对苯二酚为内标,既不干扰样品峰,又在CD3OD中有较好的溶解性。
2.1.4 1H-NMR峰的归属茶苯海明为苯海拉明与8-氯茶碱的1︰1混合物,在一张谱图中分别对2个化合物的峰进行了归属,其中归属为苯海拉明的信号峰:δ7.22~7.33(m,10H,2个苯环上的H),δ5.45(s,1H,次甲基),δ3.75(t,2H,-CH2-),δ3.39(t,2H,-CH2-),δ2.87(s,6H,2个-CH3);归属中国药事2018年6月第32卷第6期745 zhgysh CHINESE PHARMACEUTICAL AFFAIRS为8-氯茶碱的信号峰:δ3.47(s,3H,-CH3),δ3.33(s,3H,-CH3)。溶剂甲醇的信号峰分别为δ3.33与δ4.90(水峰)。
2.1.5 定量峰的选择选择苯海拉明中δ2.87的6H信号峰与8-氯茶碱δ3.47的3H信号峰作为样品定量峰,选择δ6.64的4H信号峰为内标定量峰,所选信号峰均为单峰,且互不干扰测定。见图 2。

|
图 2 茶苯海明各定量峰信号 A.内标(IS);B. 8-氯茶碱的H信号;C.苯海拉明的H信号。 |
在进行核磁定量试验时,为保证积分结果的准确,延迟弛豫时间(D1)与纵向弛豫时间(T1)的比值应≥ 5[16],通过翻转恢复试验对所有质子共振谱线的T1值分别进行测试,发现最大T1为内标δ6.64的3.996 s, 样品中δ2.89与δ3.47峰的T1分别为1.059 s与1.551 s,因此,D1值设定为20 s。
2.1.7 精密度与重复性试验取样品(原料药)溶液,连续测定5次,计算样品定量峰面积与内标峰面积比值,结果苯海拉明与8-氯茶碱的RSD分别为0.07%与0.24%。另取对照品平行制备3份样品,测得苯海拉明的含量分别为53.20%、53.40%、53.35%,RSD为0.20%;测得8-氯茶碱的含量分别为45.73%、46.27%、45.99%,RSD为0.59%。
2.1.8 线性试验精密称取茶苯海明(原料药)11.11、22.36、40.79、48.56、58.19、79.82 mg与内标10.54、10.57、10.99、10.63、10.66、10.30 mg,以不同质量比制备6份待测样品,测定1H-NMR谱。以(样品/内标)质量比为横坐标(x),NMR定量峰面积比为纵坐标(y),做线性回归,得回归方程:y 茶苯海明 =0.3484x+0.0087,y8-氯茶碱 =0.1743x+0.0088,相关系数均为0.9999,线性关系良好。
2.1.9 样品溶液稳定性取供试品(原料药)溶液,分别在配制后0、3、7 h进行测定,计算样品定量峰与内标定量峰面积比值,结果苯海拉明与8-氯茶碱的RSD分别为0.01%与0.15%,表明供试品溶液在室温放置7 h稳定。
2.1.10 测定结果分别对原料药与对照品进行测定,含量结果见表 1。
|
|
表 1 茶苯海明含量测定结果 |
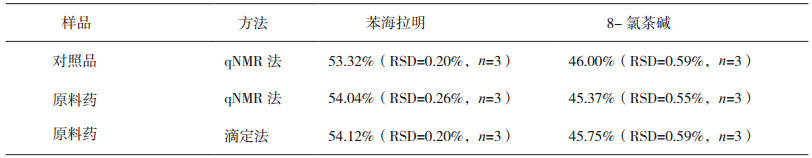
参照《中国药典》2015年版茶苯海明含量测定方法[1],采用0.1 mol·L-1高氯酸滴定液滴定苯海拉明的含量,采用0.1 mol·L-1硫氰酸铵滴定液滴定8-氯茶碱的含量,测定结果见表 1。
3 讨论目前各国药典收载的茶苯海明原料药的含量测定方法均为滴定法,样品处理与测定方法较为繁琐。由于核磁共振的原理是运用某一特定频率的电磁波来照射样品,使其原子核进行能级之间的跃迁,因此,不同化合物中氢信号的化学位移通常会有区分,本研究发挥了其在测定混合物上的技术优势,所建立的核磁定量方法,实现了在一张图谱上不仅同时测定出2种成分的绝对含量,还准确给出了2种成分含量的比例,方法准确快速,操作简单,为测定茶苯海明的含量增加了另外一种手段。
| [1] |
中国药典: 二部[S]. 2015: 713-714.
|
| [2] |
USP 38[S]. 2015: 3134.
|
| [3] |
EP 8. 0[S]. 2015: 2063-2065.
|
| [4] |
JP 17[S]. 2016: 809.
|
| [5] |
DÖGE U., EGER K.. A Simple HPLC-UV Method for the Determination of Dimenhydrinate and Related Substances-identification of an Unknown Impurity[J]. Pharmazie, 2007, 64: 174-178. |
| [6] |
Malz F., Jancke H.. Validation of Quantitative NMR[J]. J Pharm Biomed Anal, 2005, 38: 813-823. DOI:10.1016/j.jpba.2005.01.043 |
| [7] |
Holzgrabe U., Denbner R., Schollmayer C., et al. Quantitative NMR Spectroscaopy-applications in Drug Analysis[J]. J Pharm Biomed Anal, 2005, 38: 806-812. DOI:10.1016/j.jpba.2005.01.050 |
| [8] |
禹珊, 郭强胜, 王会琳, 等. 定量核磁共振波谱法同时测定中药虎杖中白藜芦醇和虎杖苷的含量[J]. 分析化学, 2015, 43(1): 69-74. |
| [9] |
胡敏, 胡昌勤, 刘文英. 核磁共振波谱法测定药物基准物质的绝对含量[J]. 分析化学, 2004, 32(4): 451-455. |
| [10] |
韦英亮, 莫建光, 潘艳坤, 等. 磺胺类标准物质期间核查NMR定量方法研究[J]. 波谱学杂志, 2013, 30(3): 371-379. |
| [11] |
Liu SY, Hu CQ. A Comparative Uncertainty Study of the Calibration of Macrolide Antibiotic Reference Standards Using Quantitative Nuclear Magnetic Resonance and Mass Balance Methods[J]. Anal Chim Acta, 2007(602): 114-121. |
| [12] |
Wu Y, He Y, He W Y, et al. Application of Quantitative 1H NMR for the Calibration of Protoberberine Alkaloid Reference Standards[J]. J Pharm Biomed Anal, 2014, 90: 92-97. DOI:10.1016/j.jpba.2013.11.018 |
| [13] |
Stephen A. Wise, Hendrik Emons.. Reference Materials for Chemical Analysis[J]. Anal Bioanal Chem, 2015, 407: 8557-8569. DOI:10.1007/s00216-015-9013-7 |
| [14] |
Michael A. Nelson, Mary Bedner, Brian E. Lang, et al. Metrological Approaches to Organic Chemical Purity:Primary Reference Materials for Vitamin D Metabolites[J]. Anal Bioanal Chem, 2015, 407: 8557-8569. DOI:10.1007/s00216-015-9013-7 |
| [15] |
中国药典: 四部[S]. 2015: 52-56.
|
| [16] |
Pauli G F, Jaki B U, Lankin D C, et al. Quantitative 1H NMR Development and Potential of an Analytical Method:An Update[J]. J Nat Prod, 2005, 68(1): 133-149. DOI:10.1021/np0497301 |