 2019, Vol. 33
2019, Vol. 33 

2. 广东省医疗器械质量监督检验所, 广州, 510663
2. Guangdong Medical Devices Quality Surveillance and Test Institute, Guangzhou 510663, China
欧洲议会和欧盟理事会发布的医疗器械法规(EU Medical Device Regulatory 2017/745)和体外诊断医疗器械监管法规(EU In Vitro Diagnostic Medical Device Regulatory 2017/746),于2019年5月开始正式实施。新法规与升级前的指令相比较,其中一个重要特点就是增强了对公告机构的监督,规范了公告机构工作程序。为了配合新法规的实施,欧盟委员会也发布了配套的执行规章(EU2017/2185)“关于欧盟议会和欧盟理事会(EU 2017/745号)法令规定的医疗器械和欧盟议会和欧盟理事会(EU 2017/746号)法令规定的体外诊断医疗器械领域的认证机构指定范围用代码和器械类别清单” [1],用于指导公告机构工作范围的准确、高效划分,促进对公告机构的科学、高效监管。认证机构指定范围用代码和器械类别清单的第一次发布是2009年,欧盟以指南的形式发布了用于指导管理机构在对公告机构工作范围进行确定过程中使用的范围清单[2],本次发布的新版本(以下简称“新框架”)是在2009年框架(以下简称“原框架”)基础上进行的改进和完善。新法规文件采取逐级细分的框架体系,将作为医疗器械监管的产品从不同角度进行了类别划分,建立了与新的法规框架下执行体系(如分类规则、命名体系)相关联和相适应的产品类别清单。
与新法规体系相适应,新框架不仅调整了结构体系,而且增加了“没有医疗预期用途的产品”等内容,为了促进中国医疗器械行业适应新形势,深度和广泛地参与全球化竞争,本文拟对新法规文件下的产品类别划分框架体系进行分析探讨,以期为建立我国医疗器械行业相关法规提供借鉴和参考。
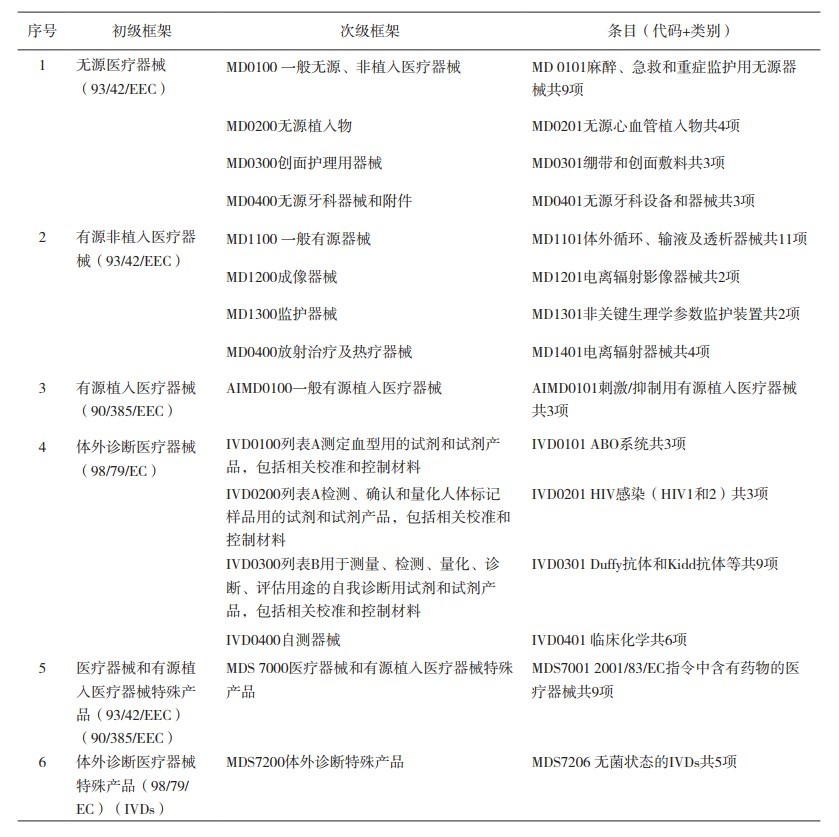
1 新旧框架基本情况 1.1 原框架简况2009年3月,欧盟发布的指南BOG - BPG 2009-3《指定机构以确定进行医疗器械评估的公告机构的通知范围的指南》(Guideline for Designating Authorities to Define the Notification Scope of a Notified Body Conducting Medical Devices Assessments)[2],在该指导原则中,将医疗器械分为无源医疗器械、有源(非植入)医疗器械、有源植入医疗器械、体外诊断医疗器械、医疗器械和有源植入医疗器械特殊产品、体外诊断医疗器械特殊产品6个初级框架,每个初级框架下分为两个层级,如表 1所示。据统计,原框架各个初级框架下条目数量为:有源医疗器械22项(有源植入3项),无源医疗器械19项,体外诊断医疗器械21项,特殊医疗器械(含有源植入器械)9项,特殊体外诊断医疗器械5项。表 1给出了公告机构医疗器械产品类别原框架基本情况。
|
|
表 1 公告机构医疗器械产品类别原框架 |
2017年发布的新框架[1],为了适应新的法规体系,在实施条例EU 2017/2185中,直接分成两部分,分别与Regulation(EU)2017/745(即MDR)和Regulation(EU)2017/746(即IVDR)相对应。第一部分具体内容:MDR对应的有源器械条目26项(有源植入4项),无源器械条目18项,横向代码条目27项;第二部分具体内容:体外诊断器械条目33项,横向代码条目47项。
1.2.1 MDR对应框架新法规的框架设置主要体现在反映器械设计和预期用途方面,基本情况如表 2所示;在设计和用途的角度之外,建立了维度来实现授权领域的准确定位,新增了横向代码设置,如表 3所示。
|
|
表 2 反映器械设计和预期用途的框架表 |

|
|
表 3 反映横向代码情况的代码表 |

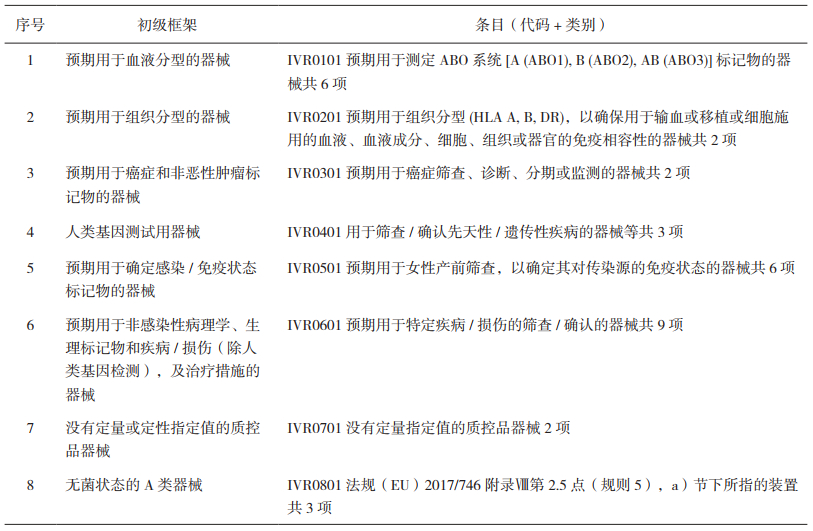
IVR体系中从反映器械设计和预期用途方面、以及从建立维度的新增横向代码两部分进行了划分,如表 4和表 5所示。
|
|
表 4 IVR体系中反映器械设计和预期用途的代码表 |
|
|
表 5 IVR体系中横向代码表 |

经对两个版本的医疗器械类别划分框架对比分析,两个框架的对应情况如下:(1)MDR对应的框架下,原版本1~3的初级框架下的条目,基本上纳入到新框架“反映器械设计和预期用途的代码”项下,初级框架5的各个条目则纳入具有特定特性的器械一项(横向代码初级框架1,见表 3)。新框架对个别条目进行了调整补充,例如,MDS1012法规(EU)2017/745附录ⅩⅥ中,新增了没有预期医疗用途的产品,体现了新法规监管思路的新动向。(2)IVDR对应的框架下,由于IVDR将之前98/79/EC的清单式分类管理系统转变为与IMDRF相接轨的分类规则制;因此,新框架反映器械设计和预期用途的代码和特性(横向代码初级框架1)相关条目按照新分类规则相关内容进行了调整和重新表述。(3)为了实现公告机构工作范围的准确划分,确保其具有相关的产品评估能力,新框架增设了横向代码,建立一个多维的产品分型系统,在产品设计和预期用途的分型角度之外,MDR框架下增设产品特定特性、特定技术工艺两个分型角度(见表 3);IVDR框架下则新增了特定特性、特定技术、所需知识等四个分型角度(见表 5),因为横向代码所选的角度都是新加入的,其项下条目也基本为新增,仅少部分来自于原框架中“特殊产品”。
为了适应法规的调整(90/385/EEC、93/42/ EEC、98/79/EC三个指令变成MDR、IVDR两个规章),有源植入器械不再作为一个单独的初级框架,而是放入了有源器械的初级框架之下。
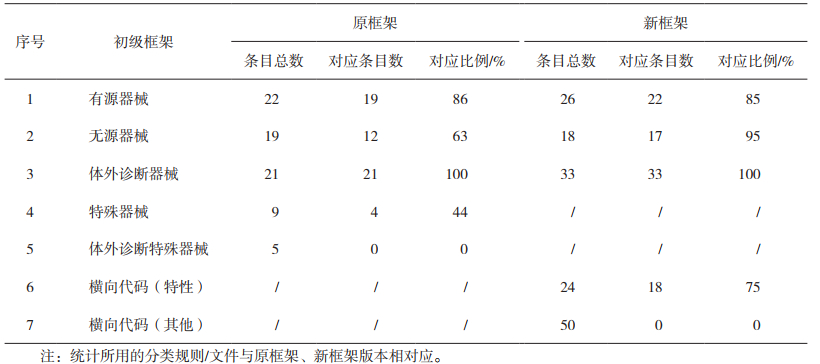
3 与EU医疗器械分类规则的关联经对比分析,为了体现和保证法规系统的一致性和完整性,不管是原框架还是新框架,相关条目的设置和表述与分类规则等管理类别文件[3-8]存在较强的对应性,例如,“有源植入器械(MDA0101~MDA0104)”4个条目均对应规则“8-植入器械和长期使用的手术侵入器械”;“成像、监测和/或诊断用有源非植入器械(MDA0201~MDA0204)”4个条目则均主要对应“规则10-预期用于诊断的有源器械”。由于分类规则与公告机构授权范围划分各自的具体适用环境和目地的差异,两者也并非完全对应,例如,“MDA 0311牙科有源非植入器械”所涉产品品类繁杂,则该条目不能直接与某个条目的分类规则相对应。横向代码中“特定特性”相关条目,主要在于体现产品特性,与分类规则有较强关联性,其他部分横向代码因为表述角度与分类规则差别大,基本上没有对应关系。对应情况统计如表 6所示。序号1~3体现的是设计和预期用途方面的特征,与基于预期用途进行风险分类的分类规则比较贴合,特别是新框架和新分类规则关联性更强,因为授权范围代码正式纳入法规框架,因此,两者的一致性显得更加必要,这有利于实际操作,利于对涉及不同风险水平产品的公告机构的有效监管,另一方面也便于公告机构采用分类规则。
|
|
表 6 公告机构医疗器械产品类别框架与分类规则对应情况统计表 |
横向代码的设置是为了在设计和用途的角度之外,另建立维度来实现授权领域的准确定位,因此,出现大幅度的不对应是合理的,体现了两个系统使用目的上的差异。
实际上,在欧盟新发布的公告机构指定范围的申请表格[9-10]中,公告机构在勾选“反映器械设计和预期用途的代码”项下条目时,还需要勾选相应附件来确定有能力进行的符合性评估程序,如表 7和表 8所示。产品适用哪些符合性评估程序,取决于其管理类别。而横向代码的勾选,有这样的设置,从另一个侧面体现了欧盟将该授权框架与分类规则相挂钩的思路。
|
|
表 7 公告机构指定范围申请表 1(“反映器械设计和预期用途的代码”部分) |

|
|
表 8 公告机构指定范围申请表 2(“横向代码”部分) |

修订后的监管法规,增强了对产品可追踪性的要求,其中一个措施就是新增了医疗器械产品命名的要求(MDR第26条,IVDR第23条)[6-7]。
2018年,医疗器械协调工作组(Medical Device Coordination Group,MDCG)代表欧盟委员会发布关于医疗器械命名的工作文件“ MDCG 2018- 2 Future EU Medical Device Nomenclature Description of Requirements” [11],其中,要求命名系统的构建设计,应有利于建立其与公告机构指定范围代码之间的联系。这一要求体现了欧盟医疗器械监管的新思路:借助新框架与命名系统挂钩,将对公告机构符合性评估活动信息纳入到欧洲医疗器械数据库(European Database on Medical Devices,Eudamed),加强医疗器械产业各个环节的全面整体监管,提升产品在各个环节的可追溯性。
5 小结欧盟医疗器械新框架以执行规章(EU)2017/2185的形式发布,与发布原框架的指南文件相比具有更强的法律效力。新框架提供了一个医疗器械的多维分型系统,使公告机构指定范围的划分更为精准合理。新框架中相关条目与分类规则、命名系统的整体化设计思路,体现欧盟监管方打造一个整体协调、高效一致的监管法规系统的工作思路。横向代码的设置,体现出监管当局不再局限于以产品风险水平作为唯一依据,对公告机构和医疗器械行业进行监管。欧盟的医疗器械管理分类体系是采用分类规则制,没有制定具体的分类目录,考虑到新框架与分类规则的对应关系,及其对命名系统的影响,这一框架在整个监管环境中的全面应用,将有助于分类规则和命名系统的落地执行,在实际操作过程中起到类似于我国分类目录的作用。我国2017年发布的分类目录框架设计上参考了欧盟原有框架[12-13],体现了中国与欧盟医疗器械监管思路的共同之处,为我国医疗器械行业参与国际化竞争提供法规方面的借鉴和参考。
| [1] |
The European commission. Commission Implementing Regulation(EU)2017/2185 of 23 November 2017 on the List of Codes and Corresponding Types of Devices for the Purpose of Specifying the Scope of the Designation as Notified Bodies in the Field of Medical Devices under Regulation(EU)2017/745 of the European Parliament and of the Council and in vitro Diagnostic Medical Devices under Regulation(EU)2017/746 of the European Parliament and of the Council[EB/OL].(2017-11-23)[2019-05-02]. https://eur-lex.europa.eu/eli/reg_impl/2017/2185/oj.
|
| [2] |
The European commission. Guideline for Designating Authorities to Define the Notification Scope of a Notified Body Conducting Medical Devices Assessments[EB/OL].(2009-03)[2019-05-02]. https://eur-lex.europa.eu/search.html?lang=en&text=a+Notified+Body&qid=1562554475649&type=quick&scope=EURLEX&DD_YEAR=2009.
|
| [3] |
The council of the European communities. Council Directive 93/42/EEC of 14 June 1993 Concerning Medical Devices[EB/OL].(2007-09-05)[2019-05-02]. https://eur-lex.europa.eu/search.html?lang=en&text=Council+Directive+93%2F42%2FEEC+of+14+June+1993+concerning+medical+devices&qid=1562554600372&type=quick&scope=EURLEX&DD_YEAR=1993.
|
| [4] |
The European parliament and the council of the European Union. Directive 98/79/EC of the European Parliament and of the Council of 27 October 1998 on in vitro Diagnostic Medical Devices[EB/OL].(2011-12-20)[2019-05-02]. https://eur-lex.europa.eu/search.html?qid=1562554770338&text=Directive%2098/79/EC%20of%20the%20European%20Parliament%20and%20of%20the%20council%20of%2027%20October%201998%20on%20in%20vitro%20diagnostic%20medical%20devices&scope=EURLEX&type=quick&lang=en.
|
| [5] |
The Council of the European Communities. Council Directive of 20 June 1990 on the Approximation of the Laws of the Member States Relating to Active Implantable Medical Devices[EB/OL].(2007-09-05)[2019-05-02]. https://eur-lex.europa.eu/search.html?lang=en&text=Council+Directive+of+20+June+1990+on+the+approximation+of+the+laws+of+the+Member+States+relating+to+active+implantable+medical+devices&qid=1562554851086&type=quick&scope=EURLEX&DD_YEAR=1990.
|
| [6] |
Council of the European Union. Regulation(EU)2017/745 of the European Parliamentand of the Council of 5 April 2017 on Medical Devices, Amending Directive 2001/83/EC, Regulation(EC)No 178/2002 and Regulation(EC) No 1223/2009 and Repealing Council Directives 90/385/EEC and 93/42/EEC[EB/OL].(2017-05-25)[2019-05-02]. https://eur-lex.europa.eu/search.html?qid=1562555005376&text=amending%20Directive%202001/83/EC,%20Regulation%20(EC)%20No%20178/2002%20and%20Regulation%20(EC)%20No%201223/2009%20and%20repealing%20Council%20Directives%2090/385/EEC%20and%2093/42/EEC&scope=EURLEX&type=quick&lang=en.
|
| [7] |
Council of the European Union. Regulation(EU)2017/746 of the European Parliament and of the Council of 5 April 2017 on in vitro Diagnostic Medical Devices and Repealing Directive 98/79/EC and Commission Decision 2010/227/EU[EB/OL].(2017-05-25)[2019-05-02]. https://eurlex.europa.eu/search.html?qid=1562555005376&text=amending%20Directive%202001/83/EC,%20Regulation%20(EC)%20No%20178/2002%20and%20Regulation%20(EC)%20No%201223/2009%20and%20repealing%20Council%20Directives%2090/385/EEC%20and%2093/42/EEC&scope=EURLEX&type=quick&lang=en.
|
| [8] |
The European commission. Guidelines on the Qualification and Classification of Stand alone Software Used in Healthcare within the Regulatory Framework of MedicalDevices[EB/OL].(2016-07)[2019-05-02]. https://eur-lex.europa.eu/search.html?qid=1562555297470&text=Guidelines%20on%20the%20qualification%20and%20classification%20of%20stand%20alone%20software%20used%20in%20healthcare%20within%20the%20regulatory%20framework%20of%20medical%20devices&scope=EURLEX&type=quick&lang=en.
|
| [9] |
The European commission. Applied for Scope of Designation and Notification of a Conformity Assessment BodyRegulation (EU) 2017/745(MDR)[EB/OL]. (2017)[2019-05-02]. https://eur-lex.europa.eu/search.html?qid=1562555513642&text=Applied-for%20scope%20of%20designation%20and%20notification%20of%20a%20Conformity%20Assessment%20body&scope=EURLEX&type=quick&lang=en.
|
| [10] |
The European commission. Applied for Scope of Designation and Notification of a Conformity Assessment Body-Regulation(EU)2017/746(IVDR)[EB/OL]. (2017)[2019-05-02]. https://eur-lex.europa.eu/search.html?qid=1562555513642&text=Applied-for%20scope%20of%20designation%20and%20notification%20of%20a%20Conformity%20Assessment%20body&scope=EURLEX&type=quick&lang=en.
|
| [11] |
Medical Device Coordination Group. MDCG 2018-2 Future EU Medical Device Nomenclature Description of Requirements[EB/OL].(2018)[2019-05-02]. https://eur-lex.europa.eu/search.html?qid=1562555806367&text=medical%20device%20nomenclature%20Description%20of%20requirements&scope=EURLEX&type=quick&lang=en.
|
| [12] |
张春青, 王越, 周良彬, 等. 我国医疗器械分类目录修订框架设计的研究[J]. 中国药事, 2017, 31(10): 1102-1106. |
| [13] |
国家食品药品监督管理总局. 2017年第104号国家食品药品监督管理总局通告[EB/OL].(2017-08-31)[2019-05-02]. http://samr.cfda.gov.cn/WS01/CL0087/17789.html.
|