 2019, Vol. 33
2019, Vol. 33 

血筛试剂是指用于血源筛查的体外诊断试剂,主要分为血清学检测和核酸检测试剂两大类。应用血筛试剂对输血相关传染病病原体进行筛查,是控制传染病疫情的重要手段,也是保障输血和血液制品安全的关键。我国纳入监管的输血相关传染病病原体包括乙型肝炎病毒(Hepatitis B Virus,HBV)、丙型肝炎病毒(Hepatitis C Virus,HCV)、人类免疫缺陷病毒(Human Immunodeficiency Virus,HIV)和梅毒螺旋体(Treponema Pallidum,TP)等。目前,只有乙型肝炎疫苗可以显著预防和控制HBV感染,而对于HCV及HIV感染,现阶段仍无有效的疫苗预防和根治手段,需用高灵敏度的血筛试剂对病原体进行筛查,以降低病原体的经血传播风险。鉴于血筛试剂的重要应用价值和巨大市场前景,血筛试剂研制一直是检验医学的热点之一。从最初的血清学检测到后来的核酸检测,血筛试剂在灵敏度、检测效率、可检测的病原体种类等方面都有显著提高。
由于血筛试剂的使用直接关系到公共健康,各国政府都对血筛试剂进行严格监管,不仅制定了相关标准及法规对其生产全过程进行质量控制,还对已上市的血筛试剂实施批签发管理,以进一步保障血筛试剂的质量,减少经输血传播的感染病例发生。我国《生物制品批签发管理办法》 [1]中明确指出,血筛试剂批签发是指原国家食品药品监督管理总局(以下简称“国家药监局”)对获得上市许可的用于血源筛查的体外诊断试剂在每批产品上市销售前或者进口时,指定药品检验机构进行资料审核、现场核实、样品检验的监督管理行为,未通过批签发的产品不得上市销售或者进口。我国于1994年开始对血清学检测血筛试剂实施批签发,对保障我国血清学检测血筛试剂的质量和临床用血安全起到了很大作用。但是,到目前为止,我国尚未对用于血源筛查用核酸检测试剂(以下简称“核酸血筛试剂”)实施批签发。
本文概述了欧盟和美国对血筛试剂批签发检验和监管的法规及实施现状,并分析我国血筛试剂批签发的检验与监管现状,为我国血筛试剂批签发监管提供参考和建议。
1 欧盟和美国血筛试剂批签发检验和监管欧盟和美国是体外诊断试剂的制造和使用大国,聚集了国际领先的血筛试剂及配套检验仪器的制造商,拥有相对完善的血筛试剂监管法规体系,积累了大量的监管经验,对各国相关法规及指导性文件的制定有重要影响。欧盟和美国对血筛试剂都进行上市前和上市后监管,对血筛试剂实施批签发管理。
1.1 欧盟的血筛试剂批签发检验和监管 1.1.1 血筛试剂批签发监管的现行法规血筛试剂在欧盟按医疗器械管理。欧盟于1998年发布了单独的医疗器械指令—《欧盟体外诊断医疗器械第98/79号指令》(European Directive In Vitro Diagnostic Medical Device Directive 98/79/EC,以下简称“IVDD”)[2],于2003年12月7日起实施。欧盟依据IVDD对体外诊断产品按风险程度高低进行分级管理。IVDD附录Ⅱ将血筛试剂列入风险最高类别的清单A类,包括HBV、HCV、HIV、丁型肝炎(Hepatitis D Virus,HDV)和人嗜T淋巴细胞病毒(Human T-cell Lymphotropic Virus,HTLV)的诊断试剂。IVDD附录Ⅳ的全面质量保证体系(EC合格声明)第6条中提出了对清单A类器械的批签发要求:制造商应在完成控制与测试之后立即向指定机构提交有关制造的器械或每批器械的测试报告;此外,制造商应按预先商定的条件和方式向指定机构提供制造的器械或每批器械的样品;指定机构需在收到样品后30天内通知制造商是否可以放行。指定机构是由欧盟各成员负责指定的第三方机构,即公告机构(Notified Body,NB)。公告机构受其所在成员国认可机构监督管理,并依据相关指令对产品执行符合性评估审核、授权CE标识(Conformité Européene Marking)使用、签发批签发报告和执行监管。
1.1.2 血筛试剂批签发检验和监管现状由于欧盟IVDD的制定是为协调各成员国的法律法规要求,因此,对批签发的规定相对简单,具有较大的灵活性,这也导致各成员国对血筛试剂批签发有各自不同的要求,主要分为3种情况:
1)英国和德国 英国和德国的公告机构(如UL、TÜV SÜD、TÜV莱茵、MDC等)的批签发都是将产品交德国血清研究所实验室(Paul-EhrlichInstitut,PEI)进行检测。在对制造商的设计文档进行审查的阶段,公告机构一方面会确认制造商自己的成品检验/放行标准是否合理,另一方面会和PEI确认批签发检验时使用的参考品。试剂产品签发前,制造商的质量部需用企业自己的参考品进行成品检验,还需将样品发送至PEI。PEI收到样品后会用事先确认好的自己研制的用于批签发检验参考品进行检验,检验结果将被直接发送到公告机构,作为公告机构同意或拒绝签发的依据。如果公告机构最终拒绝批签发,PEI的检验结果还将被告知主管当局(监管政府部门)。PEI并不销售其参考品给制造商,制造商需自己寄样品至PEI,公告机构不做抽样。
2)法国 与英国和德国不同,法国公告机构(如G-MED)的批签发是出售参考品给制造商,由制造商自检后将检验结果反馈给公告机构,公告机构据此决定是否签发批签发报告。由于公告机构出售给制造商的是一套固定的参考品,制造商实际上可以预估到参考品的检验结果,因此这种类型的批签发存在很大缺陷。
3)捷克、斯洛伐克等东欧国家 这些国家公告机构(如ITC、3EC等)的批签发采用复核企业成品检验报告的方式。公告机构确认企业成品检验流程符合规定并且成品检验结果合格,则签发批签发报告。此种方式比较粗放,也一直存在很大争议。近年来,随着欧盟对公告机构监管力度的加强,这些公告机构也在寻找一些欧洲实验室作为合作伙伴对产品进行检验,但到目前为止,尚未完成从完全不检验到要检验的转换。
1.1.3 血筛试剂批签发监管的新法规由于IVDD对批签发的规定相对简单,导致各成员国对血筛试剂批签发的流程不统一,这是目前欧盟监管面临的问题。由于公告机构的批签发报告可在欧盟各成员国通行,一旦公告机构签发批签发报告,被签发批次的产品即可在所有成员国销售。公告机构对产品批签发要求的不同,可能会导致整个欧盟市场上流通的血筛试剂产品质量良莠不齐。
为进一步统一欧盟各成员国对体外诊断试剂的监管尺度,欧盟于2017年5月5日发布了《欧盟体外诊断医疗器械第2017/746号法规》(In Vitro Diagnostic Medical Devices Regulation(EU)2017/746,以下简称“IVDR”)[3],IVDR将于2022年5月26日实施,并取代IVDD。
IVDR附录Ⅷ将血筛试剂纳入最严监管登记的D类医疗器械进行管理,并在附录Ⅸ(体外诊断企业质量保证体系的规定)对批签发流程做出新的要求:每个批次的产品需由生产商发送至指定的欧盟参考实验室(Reference Laboratory)进行检验,即批签发检验。检验结果将由参考实验室直接返回公告机构,而公告机构将根据参考实验室的检测结果以及企业自己的成品检验报告,决定是否签发批签发报告。对于高风险的血筛试剂,公告机构还需对生产者的设计文档进行审查。附录Ⅷ还对参考实验室的工作任务做出详细规定,包括对批签发产品的检验,以及向欧盟委员会和公告机构提供IVDR实施的科学和技术援助等。欧盟计划于2021底开始审批参考实验室,到目前为止,欧盟尚未有参考实验室,IVDR规定的批签发的具体操作方法尚未细化。
由于IVDR明确规定了要由参考实验室对产品进行检验,因此,IVDR实施后,欧盟各个成员国对血筛试剂批签发的流程不统一的问题将得到解决,出售参考品给制造商并由制造商反馈检验结果给公告机构以及复核企业成品检验报告的批签发方式都将不再被认可。
1.2 美国对血筛试剂批签发检验和监管在美国,血筛试剂按照生物制品管理,实施批签发。美国在批签发法规要求和监管执行上均严于欧盟。与欧盟的分权式管理不同,美国法规的执行采用集中式管理,对于血筛试剂的批签发检验和监管都由美国食品药品监督管理局(Food and Drug Administration,FDA)下属的生物制品评价和研究中心(Center for Biologics Evaluation and Research,CBER)执行。
1.2.1 血筛试剂批签发检验和监管的法规在美国,血筛试剂产品注册前需先通过生物制品许可申请(Biologics License Application,B L A)。制造商需提供3个不同批号的试剂给CBER,CBER用自己的参考品及制造商提供的3批试剂进行检验,以验证制造商的检验仪器性能。在制造商申请试剂注册时,由于检验仪器性能验证阶段已做过的血筛试剂检验,FDA便不再要求对试剂进行注册检验。美国血筛试剂批签发的法律依据是《美国联邦法规》(Code of Federal Regulations,C F R)第2 1卷“食品与药品”第6 1 0章节“通用生物制品标准”中第A分章(2 1 C F R §610.1 & §610.2)[4],明确要求每个批次生物制品放行前都要进行检验。据此,CEBR可随时要求生产商提交每一批次产品的检验结果和样品。根据CFR第21卷第610.44节要求,制造商应使用FDA提供的参考品或FDA认可的参考品,对每一次批次的产品都进行检验。如果制造商的已上市产品批签发检验一直保持质量合格,制造商也可以向FDA提出申请适当减少批签发检验频率。
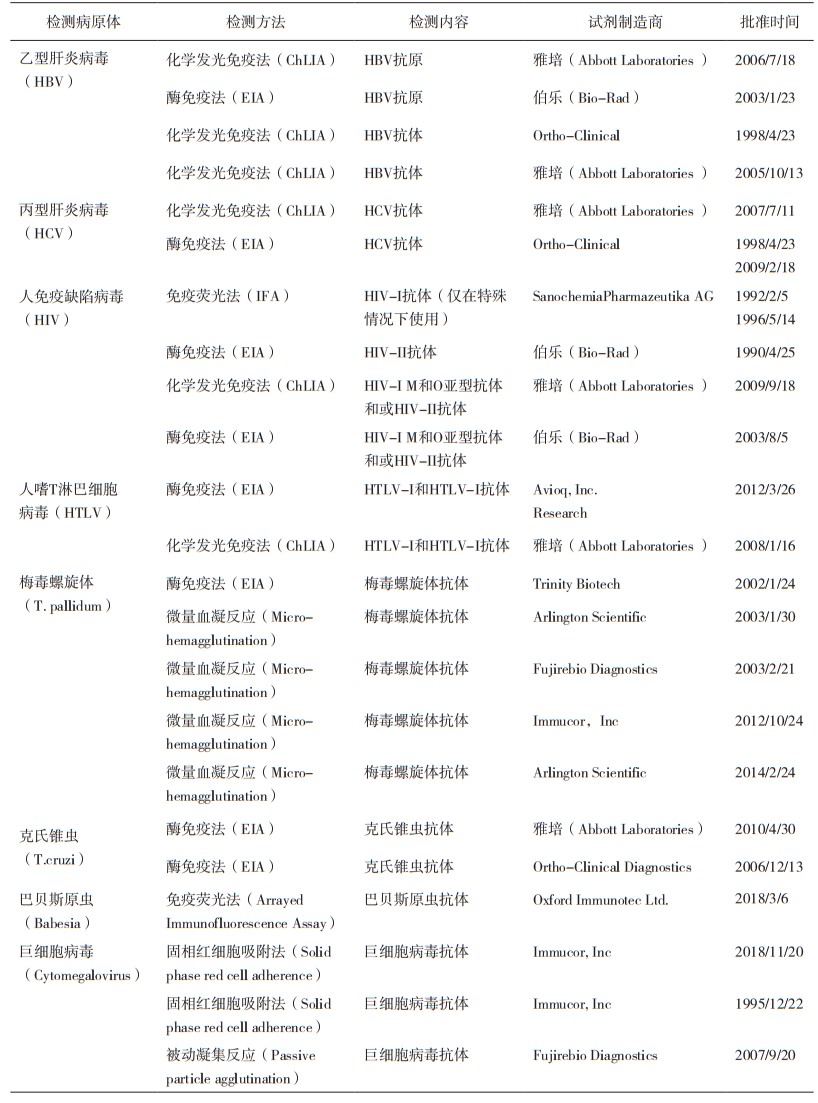
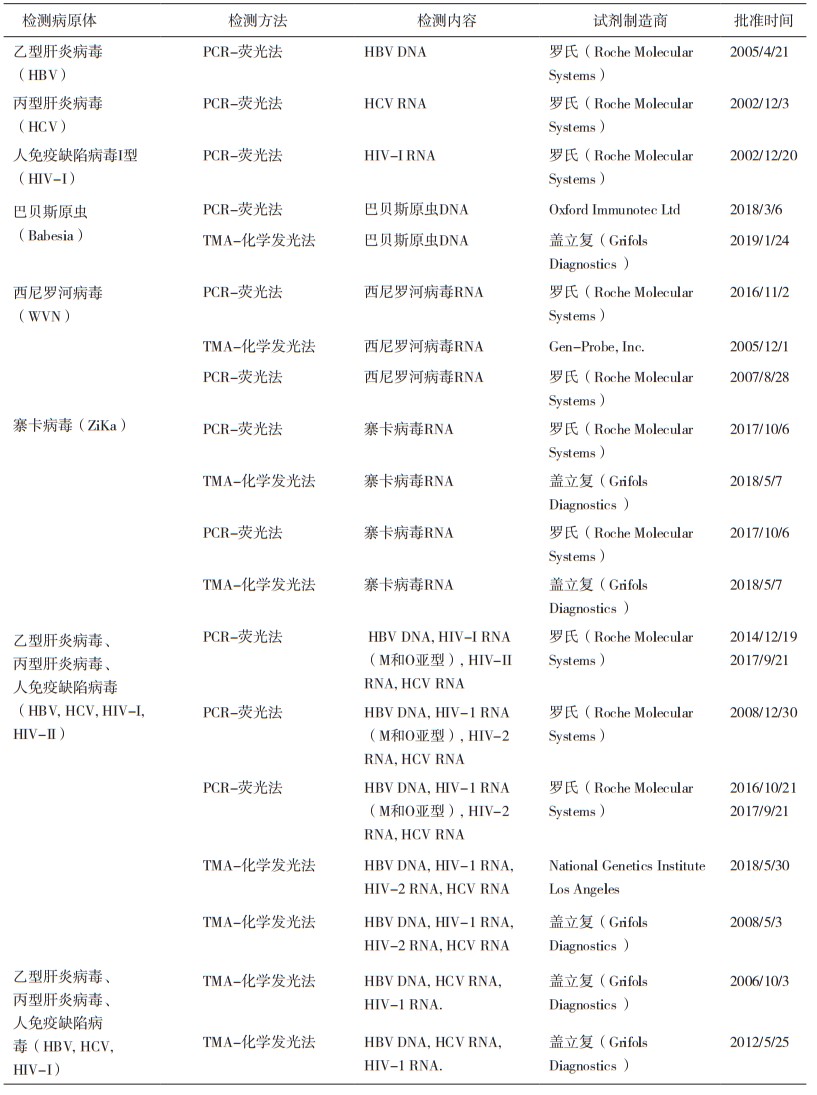
CFR没有列举具体的血筛试剂品种,在FDA网站公布的血筛试剂品种包括HBV、HCV、HIV等血清学检测试剂,以及HBV、HCV、HIV、巴贝斯原虫(Babesia)、西尼罗河病毒(West Nile Virus,WNV)和寨卡病毒(Zika)核酸检测试剂(详见表 1和表 2)。
|
|
表 1 FDA网站公布的血清学检测血筛试剂 |
|
|
表 2 FDA网站公布的核酸血筛试剂 |
C E B R下设的执法和生物制品质量办公室(Office of Compliance and Biologics Quality,OCBQ)及血液研究和审查办公室(Office of Blood Research and Review,OBRR)分别负责产品的批签发检验和签发产品放行报告工作。OCBQ还下设生产部和产品质量部(Division of Manufacturing and Quality,DMPQ)。批签发检验计划由DMPQ下属的化学、制造和控制室(Chemistry and Manufacturing Control,CMC)的主审及审批小组组长共同制定。通常制造商有企业自己的参考品,产品放行前,制造商需用自己的参考品对每一批次的产品做检验,然后将每一批次的样品发送到OCBQ下属的生物制品标准与质量控制部(Division of Biological Standards and Quality Control,DBSQC)进行检验。制造商可以向CEBR提供放行标准建议,CEBR可以接受或拒绝制造商的建议,也可以要求制造商重新修改建议。最后检验的结果将被发送到OBRR下属的新发和输血传播疾病部(Division of Emerging and Transfusion Transmitted Diseases,DETTD),由DETTD审核后决定是否签发该批次放行报告。
通常制造商需先用CBER提供的参考品进行成品检验,再送样到CEBR进行批签发检验。CEBR的检验仪器由制造商提供并由制造商负责维护保养,例如雅培(Abbott Laboratories)在美国出售的6种血清学检测试剂(详见表 1)的批签发,罗氏诊断(Roche Molecular Diagnositics)的Cobas s201检测系统配套的CobasTaqScreen MPX v2.0核酸检测试剂的批签发,及盖立复(Grifols)Panther检测系统配套的Procleix Ultrio Elite核酸检测试剂的批签发都是采用此流程。近几年,随着检测技术的发展,检测仪器的自动化程度也日益提高。针对一些大型全自动化检测仪器配套的核酸血筛试剂,例如罗氏诊断(Roche Molecular Systems)的Cobas 6800/8800检测系统配套的Cobas MPX Test核酸检测试剂,FDA的检验策略是:对每一批次的产品都提供一套专门的参考品,但并不告知企业这些参考品的具体信息,企业在此前提下做成品检验,并将检验结果的所有原始数据提交到DETTD,经由DETTD审核后决定是否放行。
2 我国血筛试剂批签发检验及监管概况与欧盟和美国类似,我国也将体外诊断试剂产品按风险不同进行分类管理。为保证我国血筛试剂的质量,《生物制品批签发管理办法》 [1]明确规定了血筛试剂按照生物制品进行管理,实施批签发。
2.1 我国对血源筛查血清学检测试剂批签发概况我国于1994年开始对血源筛查血清学检测试剂实施批签发,经过多年批签发管理,血清学检测试剂质量一直维持在较好的水平。2012-2017年,连续五年批签发的制品批次合格率保持在100%,仅2018年有1批制品不合格,1批制品的批签发资料出现质量授权人变更和关键原材料来源发生变化的问题,联合辖区药监部门一起开展了现场核实[5]。已开展的血清学检测试剂批签发工作对保障我国临床用血安全起到很大作用,世界卫生组织(World Health Organization,WHO)发布的《2016年全球血液安全与供应报告》 [6]也显示,中国在保障血液安全供应和临床合理用血等方面成就巨大,居全球前列。
2.2 我国对核酸血筛试剂的监管概况到目前为止,我国尚未开展核酸血筛试剂的批签发检验。但是,核酸检测试剂在我国上市前,国家药监局药品评审中心对试剂都有严格的质量评价,只有通过了严格的注册检验、审评审批和临床验证的试剂才可获批上市。因此,已获批的核酸检测试剂都应符合国家标准和产品注册标准,并达到相应的质量要求。而且,进口核酸检测试剂有进口检验,据文献[7]报道国产的与进口的核酸检测试剂在检出性能上没有显著差异。
3 对我国血筛试剂批签发检验和监管思路的建议《生物制品批签发管理办法》 [1]第二章第七条规定批签发机构及其所负责的批签发品种由国家药监局确定,国家药监局根据批签发工作需要,适时公布新增批签发机构及批签发机构扩增批签发品种的遴选标准和条件。
3.1 对我国血筛试剂监管品种遴选的建议我国流行病学调查表明HBV、HTLV等流行分布具有年龄和地域差异性[8]。例如HTLV在我国多数地区低流行,但在福建、广东等沿海地区高度流行[9],而来自这些HTLV高度流行的福建、广东等沿海地区的献血人群比例日渐上升[10]。尽管现行《血站技术操作规程(2015年版)》(以下简称《规程(2015年版)》)[2]及2019年9月1日即将实施的《血站技术操作规程(2019年版)》(以下简称《规程(2019年版)》)[3]均提出,血液检测项目包括国家和省级卫生计生行政部门规定的地方性、时限性输血相关传染病标志物,但是,目前我国只有HBV、HCV、HIV的血清学检测试剂被纳入批签发,HTLV的血清学血筛试剂尚未纳入批签发。因此,建议国家药监局适时增加HTLV的血清学检测试剂等地方性、时限性输血相关传染病标志物的检测试剂批签发项目。
3.2 对开展核酸血筛试剂批签发检验的建议尽管血清学检测试剂在血液筛查中起到了很好的作用,但病毒感染检测的“窗口期”依然是世界难题,对于“窗口期”病原体感染及隐匿性病原体感染,单纯抗原或抗体的检测并不能完全保障输血和血液安全。文献[11]报道,应用核酸检测试剂进行血源筛查,可以进一步缩短“窗口期”,提高病毒检出率,可降低因隐匿性病毒感染、窗口期等因素造成的漏检[12-13]。而且分子流行病学研究[4]表明,我国常见肝炎病毒和艾滋病病毒基因重组频繁,种类复杂且多样,传播网络不断扩大,应用核酸检测可以区别不同基因型别病毒感染,尽早诊断出阳性感染者。
一些发达国家如日本、美国、德国等,早已将核酸检测试剂应用到血液初筛中[14]。WHO在2009年发布的《血液筛查建议书》 [15]对艾滋病病毒核酸检测提出指导:许多国家已经实行艾滋病病毒RNA筛查;艾滋病病毒RNA是艾滋病病毒感染后最先出现在血循环中的标志物,从检出艾滋病病毒RNA到检出艾滋病病毒P24抗原之间的窗口期可能较短,艾滋病病毒RNA筛查价值与所使用的血清学筛查试验以及献血者艾滋病病毒感染发生率有关。
为进一步提高临床用血质量,我国卫生部在2000年开展了核酸检测用于血源筛查的试点工作。2012年10月,国务院发布了《卫生事业“十二五”发展规划》,指出到“十二五”末,血液筛查核酸检测技术将基本覆盖全国[16]。原国家卫生和计划生育委员会在2015年12月发布的《规程(2015年版)》将核酸检测作为血液检测方法之一,提出实施核酸检测试剂批签发之后,HIV、HBV和HCV感染标志物应采用核酸和血清学检测2种方法各进行1次检测[17]。在检测试剂选择中强调了必须选择经国家药品监督管理部门批准用于血源筛查的体外诊断试剂。在试剂质量控制的条款中规定对于血源筛查的体外诊断试剂盒,必须有国家批批检报告。由于到目前为止,我国尚未开展核酸血筛试剂批签,因此,《规程(2019年版)》 [18]提出,如果检测项目中所需的体外诊断试剂无经国家药品监督管理部门批准用于血源筛查的,则可选择经过批准的体外诊断试剂。因此,建议国家药监局加快推动落实核酸血筛试剂批签发工作,以便采(供)血站有经过批签发检验的核酸检测试剂可用。
3.3 对我国核酸血筛试剂批签发检验模式的建议若开展核酸血筛试剂的批签发检验工作,建议借鉴欧盟和美国血筛试剂批签发监管经验,综合评价核酸血筛所能提高的血液安全程度和增加的冷链运输费用及检测费用,制定适合我国国情的批签发检验模式。
核酸血筛试剂批签发如果采用给企业发参考品进行检测,监管机构进行结果审核的方式,则需要另外建立一套新的批签发系统,其中存在的风险在现阶段还无法预知。而由企业自检,监管机构复核企业成品检验报告的批签发方式,在国际上争议最大,现阶段我国也不适用。
从国际监管趋势看,批签发均走向全面检验之路。鉴于现阶段我国部分企业的诚信度还有待提高,同时,目前实施的血清学血筛试剂批签发监管和检验系统是经过多年实践检验的,从样品的收取到检验报告的发出,及后续问题处置的各环节都已相对完善。若开展核酸血筛试剂批签发工作,建议参照血清学检测试剂批签发模式,由国家药监局指定检验机构开展全部项目检验。建议国家药监局在遴选核酸检测试剂检验机构时,充分考虑检验团队的专业程度、稳定性和检验人员的专业素质、技术搭配及检验经验等因素。由于现阶段省级药品检定所均未开展诊断试剂检验工作,不具备相关的工作经验及仪器设备,建议今后可以再根据实际情况,在个别地区布点,适当扩大授权。
4 结语我国对血清学检测试剂具有良好的批签发管理,血清学检测试剂一直保持较高的批签发合格率。虽然核酸血筛试剂目前尚未实施批签发管理,但是有严格的上市前质量控制,对于保障我国的血液和血制品安全起到了很大作用。随着我国经济的发展,旅游活动、国际交流和劳务输出等引发的输入性传染病也不断增多,传播网络不断扩大。而且,我国是人口大国,地域辽阔,病原体流行株的种类更加复杂,不仅存在一些特有的病毒亚型,病原体在我国不同地区的流行趋势也有差异,需要同时应用核酸检测和血清学检测对血源进行筛查,最大限度地提高病原体检出率。为保障血筛试剂的质量,我国应制定符合国情的、灵活有效的血筛试剂批签发检验及监管政策。另外,还应该看到,单纯依靠血筛试剂质量控制并不能完全保障用血安全,最主要的还需血站和浆站建立完善的质量体系。因此,血站和浆站应对影响血液检测的关键点进行全面的质量管理[19],以保证过程管理的有效性和最终保障供血的安全性。
| [1] |
国家食品药品监督管理总局.国家食品药品监督管理总局第39号令生物制品批签发管理办法[S]. 2017.
|
| [2] |
Europe C o. European Directive in Vitro Diagnostic Medical Device Directive 98/79/EC(IVDD)[S]. 1998.
|
| [3] |
Europe C o. in Vitro Diagnostic Medical Devices Regulation (EU)2017/746(IVDR)[S]. 2017.
|
| [4] |
Department of Health and Human Services F. 21 CFR Parts 606, 607, 610, 640, 660, and 809[J]. Federal Register, 2001, 66(112): 31146-31165.
|
| [5] |
中国食品药品检定研究院. 2018年生物制品批签发年报[R]. 2019.
|
| [6] |
世界卫生组织(WHO). 2016年全球血液安全与供应报告[R]. 2017.
|
| [7] |
梁启忠, 程玉根. 核酸检测技术在献血者血液筛查中的应用[J]. 国际检验医学杂志, 2015, 36(22): 3265-3267, 3270. DOI:10.3969/j.issn.1673-4130.2015.22.018 |
| [8] |
刘纯, 刘伟, 王凯, 等. 天津地区慢性乙型肝炎患者病毒基因型的分析及临床意义[J]. 中华医院感染学杂志, 2014, 24(10): 2399-2401. |
| [9] |
季阳, 苑宇哲, 蔡辉, 等. 输血传播人类嗜T淋巴细胞病毒感染及其预防对策[J]. 中国输血杂志, 2010, 23(12): 1003-1005. |
| [10] |
张钢, 蒋义. 长沙地区献血人群HTLV感染情况调查与策略研究[J]. 实用预防医学, 2018, 25(7): 826-828. DOI:10.3969/j.issn.1006-3110.2018.07.016 |
| [11] |
Roth WK B S, Drosten C, et al. NAT and Viral Safety in Blood Transfusion[J]. Vox Sanguinis, 2000, 78(2): 257-259. |
| [12] |
赵红娜, 王艺芳, 葛文超, 等. 郑州地区无偿献血者中核酸检测的初步应用[J]. 中国输血杂志, 2014, 27(9): 923-926. |
| [13] |
曹华琳, 刘亚军. 核酸检测与酶联免疫检测对输血相关传染性疾病的检测效果对比分析[J]. 心脑血管病防治, 2019, 19(2): 171-173. DOI:10.3969/j.issn.1009-816x.2019.02.021 |
| [14] |
Roth W K. History and Future of Nucleic Acid Amplification Technology Blood Donor Testing[J]. Transfus Med Hemother, 2019, 46(2): 67-75. DOI:10.1159/000496749 |
| [15] |
郭永建, 池泉, 涂东晋, 等. WHO血液筛查建议书主要内容介绍[J]. 中国输血杂志, 2010, 23(1): 66-72. |
| [16] |
国务院.卫生事业发展"十二五"规划[S]. 2012.
|
| [17] |
国家卫生和计划生育委员会.血站技术操作规程(2015版)[S]. 2015.
|
| [18] |
国家卫生健康委.血站技术操作规程(2019版)[S]. 2019.
|
| [19] |
赵兰兰. 血站血液检测关键点的控制与质量管理[J]. 中国医药指南, 2018, 16(34): 294-295. |