 2019, Vol. 33
2019, Vol. 33 

2. 中国食品药品检定研究院, 北京 100050
2. National Institutes for Food and Drug Control, Beijing 100050, China
微生物污染是影响药品质量,引发药品安全问题的重要原因。据文献[1]报道,2004-2011年间,因微生物污染被美国FDA召回的事件共642起,在2010-2011年间召回数量明显增加;这些召回事件中涉及到的产品类型主要包括医疗器械,其次是药品,其中药品占比26%,包括无菌药品及非无菌药品。因此,制药环境良好的微生物控制是保证药品质量安全的重要措施。
对制药洁净环境污染微生物进行良好的控制,首先需要了解其污染微生物的组成、分布规律及特点,以便于更好地控制洁净区人员数量、规范人员更衣进场程序和洁净区行为、制定有效的清洁消毒程序,从而使洁净区处于正常的运行和良好的受控状态下,最大程度地降低药品污染微生物的可能性。
目前,制药行业洁净区微生物群落分布规律的相关研究较少,人们对制药环境了解不多[2-3]。吴根福等[4]通过对洁净室空气、表面和人员中的细菌进行长期监测,分析洁净室微生物群落的构成及筛选出优势菌株,用于研究更有效的消毒方法。国外多篇研究[2-3, 5]通过对不同洁净等级的生产环境长期监测,揭示洁净室微生物群落的分布特征,为质量管理人员提供可靠的数据参考。
本研究通过采集A、B、C、D、E五家制药企业C级和D级区的144株环境菌株,采用16S rRNA序列分析、ITS序列分析或依据种属信息增加看家基因的方法,对搜集获得的环境菌株进行鉴定。依据鉴定结果,分析制药企业环境菌的组成、分布规律及特点,为制药企业洁净环境良好的微生物控制提供数据支持。
1 仪器与试剂T100型PCR仪(BIO-RAD),NU-543-600S型生物安全柜(NUAIRE),INC821C型恒温培养箱(Yamato),SQ810C型压力灭菌器(Yamato),SCIENTZ-48型高通量组织研磨器(宁波新芝生物科技有限公司),TW12型恒温水浴(Julabo),氯仿、异戊醇、异丙醇、无水乙醇(均为国药化学试剂有限公司),PrepGEM Bacteria PBA0100型DNA提取试剂盒(ZyGEM),2×Taq Plus PCR MasterMix(TIANGEN),胰酪大豆胨琼脂培养基(Tryptic Soy Agar,TSA)(北京三药科技开发公司),沙氏葡萄糖琼脂培养基(Sabouraud Dextrose Agar Medium,SDA)(北京三药科技开发公司)。
2 方法及结果 2.1 采样与培养本研究通过沉降菌采集方式对A、B、C、D、E五家制药企业的生产洁净车间包括人员气锁、卸载间、灌装间、洁净实验室、操作人员等多个监控点采集环境菌株。
使用TSA平板,依据GB/T16294-2010《医药工业洁净室(区)沉降菌的测试方法》,按采样点布置图逐一放置,从里到外逐个打开培养皿盖,暴露于空气中30 min达到规定的时间后盖上培养皿盖子,于32.5℃培养箱中倒置培养48 h。挑取平板中菌落,分别进行2次以上TSA或SDA平板划线分离纯化菌落,用于微生物鉴定,并保存。
2.2 菌种保存用接种环挑取分离纯化的菌落置25%甘油管中,沿壁打散,涡旋至均匀的菌悬液,置于-50℃冰箱冻存。
2.3 微生物鉴定 2.3.1 基因组提取依据细菌基因组试剂盒产品说明书提取细菌基因组。采用十六烷基三甲基溴化铵法(CTAB)提取真菌基因组。
2.3.2 基因片段扩增 2.3.2.1 扩增引物本研究所用基因片段扩增引物见表 1。
|
|
表 1 基因片段扩增引物 |

16S rRNA:DNA模板1.5 μL,引物各5 pmol,2×Taq DNA聚合酶12.5 μL,用双蒸水补齐体系至25 μL。
dnaJ:DNA模板1.5 μL,引物各5 pmol,2×Taq DNA聚合酶12.5 μL,用双蒸水补齐体系至25 μL。
ITS:DNA模板2.5 μL,引物各5 pmol,2×Taq DNA聚合酶12.5 μL,用双蒸水补齐体系至25 μL。
2.3.2.3 PCR反应程序16S rRNA:95 ℃预变性,4 min;95 ℃变性,30 s,50 ℃退火,45 s,72 ℃延伸,1.5 min,循环35次;72 ℃延伸,5 min;4 ℃暂存。
dnaJ:94 ℃预变性,3 min;94 ℃变性,30s,45 ℃退火,30 s,72 ℃延伸,60 s,循环5次;94 ℃变性,30 s,50 ℃退火,30 s,72 ℃延伸,60s,循环30次;72 ℃延伸,3 min;4 ℃暂存。
ITS:95 ℃预变性,4 min;95℃变性,30 s,48 ℃退火,30 s,72 ℃延伸,1 min,循环30次;72 ℃延伸,5 min;4 ℃暂存。
2.3.3 PCR产物测序及分析PCR产物测序交由北京诺赛基因组研究中心有限公司完成,所得拼接结果输入至BLAST数据库比对。
2.4 制药企业洁净区环境微生物多样性分析本研究收集获得的144株环境菌中,16S rRNA序列分析结果判定葡萄球菌属40株,占比27.8%;微球菌属27株,占比18.8%;芽孢杆菌属16株,占比11.1%;微杆菌属9株,占比6.3%;不动杆菌属8株,占比5.6%;假单胞菌属6株,占比4.2%;真菌5株,占比3.5%;其他分属于10个属的菌株共33株,占比22.9%。见图 1。

|
图 1 五家制药企业洁净区微生物群落组成图 |
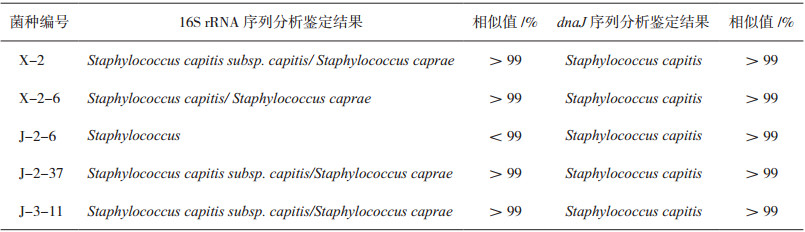
搜集到的139株细菌中,16S rRNA序列分析法能鉴定到属水平的占比100%,葡萄球菌属菌株鉴定到种水平的占比85%,芽孢杆菌属鉴定到种水平的占比43.75%,微球菌属鉴定到种水平的占比51.85%。本研究运用dnaJ基因序列分析对16S rRNA未鉴定到种的葡萄球菌属的5株菌分别进行鉴别,鉴别结果由表 2可知,对于X-2、X-2-6、J-2- 6、J-2-37、J-3-11五株菌,16S rRNA序列分析只能将其鉴定到属的水平,增加dnaJ基因可将其区分至种水平。
|
|
表 2 葡萄球菌属分离菌16S rRNA和dnaJ基因序列分析结果 |
影响洁净区微生物群落分析的重要因素包括两个方面。一是洁净区的类型和位置,二是对搜集获得的环境菌的鉴定方法[5]。洁净室的类型因需处理的产品类型不同而在设计功能上有所不同。所选洁净区是否为符合药品生产质量管理规范(Good Manufacturing Practices,GMP)的A、B、C、D洁净等级区域,洁净室的温湿度不同,出席人数不同,是否有水系统都将对搜集的微生物的种类和数量产生重要影响。微生物鉴定方法的类型及数据库的大小也会对洁净区微生物群落分析产生影响。
本研究选取的五家制药企业均符合GMP生产车间要求,洁净分区明确,同处北京,温湿度差异较小。收集菌株数量达144株,具有一定的代表性。微生物鉴定方法选择16S rRNA序列分析、ITS序列分析,同时结合看家基因序列分析,这些方法属于基因型分析方法,准确率高,数据库大,鉴定结果可靠。
3.2 制药企业洁净区微生物群落分析本研究发现,制药企业洁净室分布占比最高的菌群是革兰氏阳性球菌,包括微球菌属和葡萄球菌属,总占比46.6%;其次是革兰氏阳性杆菌,如芽孢杆菌属占比11.1%,微杆菌属占比6.3%;还有少量的革兰氏阴性杆菌如假单胞菌属、不动杆菌属总占比9.8%;真菌占比3.5%。分布特点与国外环境监测相关报道[2, 5]基本一致,以葡萄球菌属和微球菌属为主,总占比40%;其次是芽孢杆菌属,占比10%;少量的假单胞菌属,占比8%;真菌占比3%。
3.3 制药企业洁净区污染源分析正常运行的制药行业洁净区主要污染来源有四种途径:人、空气、表面及水[5]。在制药工业中最常见的微生物群落很难找到确切的来源[2]。葡萄球菌属和微球菌属等革兰氏阳性球菌,主要来源于实验室操作人员,可能是与人员皮肤脱落片状物有关[5-6]。芽孢杆菌属广泛分布于土壤中,这些微生物可能由灰尘、物料、人员转移至制药企业洁净室[5]。不动杆菌属、假单胞菌属等革兰氏阴性杆菌,可能是通过使用纯化水进行日常清洁的过程中引入[5]。假单胞菌属条件致病微生物,容易分散附着于潮湿表面,形成生物膜,干扰工业环境清洁消毒程序的进行[7],该类微生物的污染,需质量控制管理人员寻找合适有效的消毒方法。
3.4 制药企业洁净区环境菌的鉴定制药行业环境菌长期处于清洁消毒环境中,代谢活动被抑制,生化反应不典型,传统的生化鉴定方法不适用于其鉴定分析[8]。细菌的16S rRNA基因既高度保守,又相对可变,序列同源性可用于区分鉴定微生物的种类,反映微生物的进化信息[9]。16S rRNA基因序列分析也成为鉴定细菌或构建细菌系统发育关系最常用的方法[10]。通过本研究发现,从属水平上,16S rRNA序列分析法对制药环境分离菌株鉴别的准确率达到100%。从种水平上来看,在制药企业洁净室分布占比最高的葡萄球菌属、微球菌属等革兰氏阳性球菌鉴别的准确率均大于50%,说明该法可有效区分制药工业大部分环境菌,但对于部分亲缘关系较近的物种,16S rRNA基因序列分析的准确度有限[11-12]。
近年来,利用蛋白编码基因作为系统发育标记也成为常用的方法之一[13]。gyrB基因编码DNA螺旋酶的亚单位B蛋白,是一种Ⅱ型DNA拓扑异构酶,在DNA复制中起着重要作用,在细菌物种中普遍分布[14]。已有文献报道,gyrB基因可用于假单胞菌属、分枝杆菌属、沙门氏菌属、志贺氏菌属、埃希氏菌属及炭疽芽胞杆菌-苏云金芽孢杆菌属的菌种鉴定。这些研究的结果表明,gyrB是研究物种系统发育和分类关系的一个合适标记[15-21]。dnaJ,又称Hsp40,是热休克蛋白(Heat Shock Protein,Hsp)家族的成员之一,广泛分布于真核生物细胞、细菌和古细菌中[22]。有相关研究表明dnaJ基因序列可能比其他保守基因(包括16S rRNA基因)在亲缘关系较近的物种识别中更适合作为靶序列[23-24]。如果企业需要对洁净室微生物多样性进行深度分析,可针对不同种属细菌,增加看家基因,结合多种分析手段,综合分析鉴定结果。
3.5 建议综上所述,人源常见的微生物如葡萄球菌属、芽孢杆菌属、微球菌属是制药企业洁净区污染的主要来源,这与国外相关报道基本一致[2-5],再次说明人是制药行业洁净区的主要污染源;但假单胞菌属和不动杆菌属等革兰氏阴性杆菌占比明显,是由于本研究是针对于C、D低洁净等级且采用纯水进行日常清洁的区域进行微生物多样性分析。这些菌株的出现,也提示制药企业对于低等级洁净室应加强管理,寻找合理有效的清洁消毒方式,以免该类微生物转移至高等级洁净区域,从而影响终产品的安全性。
该项研究结果提示制药企业,可以使用限制性进入系统或隔离系统以部分或完全隔离人员;控制洁净区人员数量,尽可能减少人员的流动;规范人员更衣进场程序和洁净区行为,例如:进入洁净区的人员应穿着与洁净度要求相适应的防护服,佩戴帽子和口罩;洁净区内应避免跑动和快速走动;清洁或消毒洁净区时,应从高洁净等级区域逐渐过渡至低等级区域。
对洁净区内仪器设备的表面、墙面和地面应制定科学有效的消毒程序,中效消毒剂如酸性苯酚、碱性苯酚、异丙醇等,与杀孢子剂如过氧化氢、过氧乙酸等交叉使用,最大程度减少微生物污染的可能性。
制药用水是满足实验需要和人员、设备清洁所必须的物质。要密切监控纯化水或注射用水的微生物负载水平;在设备管道、洁净室地板或表面出现水溢出或长时间有水滞留的地方会滋生细菌,应及时清除,保持干燥。且注意在清洁或配制消毒剂时,应使用与洁净等级相适应的水。
制药企业需对洁净室持续进行环境监测,定期分析监控数据,研究数据变化趋势,一旦出现异常数据应及时采取措施,确认洁净区是否处于良好的受控状态。例如洁净区内突然发现大量革兰氏阴性杆菌,需要考虑是否在洁净室内有水的残留,或者是否与洁净室内湿度突然上升有关。
| [1] |
Sutton S, Jimenez L. A Review of Reported Recalls Involving Microbiological Control 2004-2011 With Emphasis on FDA Considerations of Objectionable Organisms[J]. American Pharmaceutical Review, 2012, 15(1): 42-57. |
| [2] |
Ashour M S E-D, Mansy M S, Eissa M E. Microbiological Environmental Monitoring In Pharmaceutical Facility[J]. Egyptian Academic Journal of Biological Sciences, 2011, 3(1): 63-74. DOI:10.21608/eajbsg.2011.16696 |
| [3] |
Park H K, Han J H, Joung Y, et al. Bacterial Diversity in the Indoor Air of Pharmaceutical Environment[J]. Journal of Applied Microbiology, 2014, 116(3): 718-727. DOI:10.1111/jam.2014.116.issue-3 |
| [4] |
Wu G F, Liu X H. Characterization of Predominant Bacteria Isolates from Clean Rooms in a Pharmaceutical Production Unit[J]. Journal of Zhejiang University SCIENCE B, 2007, 8(9): 666-672. DOI:10.1631/jzus.2007.B0666 |
| [5] |
Sandle T. A Review of Cleanroom Microflora:Types, Trends, and Patterns[J]. PDA Journal of Pharmaceutical Science and Technology, 2011, 65(4): 392-403. DOI:10.5731/pdajpst.2011.00765 |
| [6] |
Dao H, Lakhani P, Police A, et al. Microbial Stability of Pharmaceutical and Cosmetic Products[J]. AAPS PharmSciTech, 2018, 19(1): 60-78. DOI:10.1208/s12249-017-0875-1 |
| [7] |
Wirtanen G, Salo S. Disinfection in Food Processing Efficacy Testing of Disinfectants[J]. Reviews in Environmental Science and Bio/Technology, 2004, 2: 293-306. |
| [8] |
Jeanne E Moldenhauer A A, Susan E Arrigoni. Technical Report No.13 Revised Fundamentals of an EM Program[J]. PDA Journal of Pharmaceutical Science and Technology, 2001, 55: 1-5. |
| [9] |
Petti C A, Polage C R, Schreckenberger P. The Role of 16S rRNA Gene Sequencing in Identification of Microorganisms Misidentified by Conventional Methods[J]. Journal of Clinical Microbiology, 2005, 43(12): 6123-6125. DOI:10.1128/JCM.43.12.6123-6125.2005 |
| [10] |
P Vandamme, B pot M G, P de vos, et al. Polyphasic Taxonomy, A Consensus Approach to Bacterial Systematics[J]. Microbiological Reviews, 1996, 60(2): 407-438. |
| [11] |
Becker K, Harmsen D, Mellmann A, et al. Development and Evaluation of a Quality-Controlled Ribosomal Sequence Database for 16S Ribosomal Dna-Based Identification of Staphylococcus Species[J]. Journal of Clinical Microbiology, 2004, 42(11): 4988-4995. DOI:10.1128/JCM.42.11.4988-4995.2004 |
| [12] |
Shah M M, Iihara H, Noda M, et al. dnaJ Gene Sequence-Based Assay for Species Identification and Phylogenetic Grouping in the Genus Staphylococcus[J]. International Journal of Systematic and Evolutionary Microbiology, 2007, 57(1): 25-30. DOI:10.1099/ijs.0.64205-0 |
| [13] |
Zeigler D R. Gene Sequences Useful for Predicting Relatedness of Whole Genomes in Bacteria[J]. International Journal of Systematic and Evolutionary Microbiology, 2003, 53(6): 1893-1900. DOI:10.1099/ijs.0.02713-0 |
| [14] |
Huang W M. Bacterial Diversity Based on Type Ⅱ DNA Topoisomerase Genes[J]. Annual Review of Genetics, 1996, 30: 79-107. DOI:10.1146/annurev.genet.30.1.79 |
| [15] |
Yamamotot S, Harayama S. Phylogenetic Relationships of Pseudornonas Putida Strains Deduced from the Nucleotide Sequences of gyrB, rpoD and 16S rRNA Genes[J]. International Journal of Systematic Bacteriology, 1998, 48: 813-819. DOI:10.1099/00207713-48-3-813 |
| [16] |
Yamamoto S, Bouvet P J M, Harayamal S. Phylogenetic Structures of the Genus Acinetobacter Based on gyrB Sequences:Comparison with the Grouping by DNADNA Hybridization[J]. International Journal of Systematic Bacteriology, 1999, 49: 87-95. DOI:10.1099/00207713-49-1-87 |
| [17] |
Niemann S, Harmsen D, Sabine Rusch-gerdes, et al. Differentiation of Clinical Mycobacterium Tuberculosis Complex Isolates by gyrB DNA Sequence Polymorphism Analysis[J]. Journal of Clinical Microbiology, 2000, 38(9). |
| [18] |
Fukushima M, Kakinuma K, Kawaguchi R. Phylogenetic Analysis of Salmonella, Shigella, and Escherichia Coli Strains on the Basis of the gyrB Gene Sequence[J]. Journal of Clinical Microbiology, 2002, 40(8): 2779-2785. DOI:10.1128/JCM.40.8.2779-2785.2002 |
| [19] |
Yanez M A, Catalan V, Apraiz D, et al. Phylogenetic Analysis of Members of the Genus Aeromonas Based on gyrB Gene Sequences[J]. International Journal of Systematic and Evolutionary Microbiology, 2003, 53(3): 875-883. DOI:10.1099/ijs.0.02443-0 |
| [20] |
La Duc M T, Satomi M, Agata N, et al. gyrB as a Phylogenetic Discriminator for Members of the Bacillus Anthracis-cereus-thuringiensis Group[J]. Journal of Microbiological Methods, 2004, 56(3): 383-394. DOI:10.1016/j.mimet.2003.11.004 |
| [21] |
Wang L T, Lee F L, Tai C J, et al. Comparison of gyrB Gene Sequences, 16S rRNA Gene Sequences and DNADNA Hybridization in the Bacillus Subtilis Group[J]. International Journal of Systematic and Evolutionary Microbiology, 2007, 57(8): 1846-1850. DOI:10.1099/ijs.0.64685-0 |
| [22] |
J L Macario A, B Dugan C, Clarens M, et al. dnaJ in Archaea[J]. Nucleic Acids Research, 1993, 21(11): 2773. DOI:10.1093/nar/21.11.2773 |
| [23] |
Itoh Y, Kawamura Y, Kasai H, et al. dnaJ and gyrB Gene Sequence Relationship Among Species and Strains of Genus Streptococcus[J]. Systematic and Applied Microbiology, 2006, 29(5): 368-374. DOI:10.1016/j.syapm.2005.12.003 |
| [24] |
Das S, Dash H R, Mangwani N, et al. Understanding Molecular Identification and Polyphasic Taxonomic Approaches for Genetic Relatedness and Phylogenetic Relationships of Microorganisms[J]. Journal of Microbiological Methods, 2014, 103: 80-100. DOI:10.1016/j.mimet.2014.05.013 |