 2019, Vol. 33
2019, Vol. 33 

研究者是临床试验的实施者[1],直接影响临床试验质量和受试者权益[2]。《美国联邦法规汇编》第21篇第312.60、812.100部分条款(21 CFR 312.60、21 CFR 812.100)阐述了研究者总体职责,其定义为负责确保根据签署的研究者声明、研究计划和适用的法规开展研究,保护受试者的权利、安全、福利,以及管理试验产品[3]。警告信是美国食品药品监督管理局(FDA)发现一项或多项产品、实践、流程或其他活动违反了《美国联邦法规汇编》“食品与药品”篇相应条款后,首次发给组织或个人的正式通知[4-5]。FDA对临床试验现场检查后,若发现研究者违反了《美国联邦法规汇编》相应条款,则给研究者发警告信[6-7]。研究者自收到警告信的15个工作日内,应书面说明纠正和防止类似违规情况发生而采取的措施。如果FDA认可研究者即将或正在采取合理的纠正措施,则发终止信;如果不认可或警告信在本质上无法纠正,则不发终止信[8]。FDA给研究者发警告信的形式,有助于研究者了解临床试验的常见违规行为,尽可能避免合规性问题,也给其他国家提供了临床试验监管新思路。
我国原国家食品药品监督管理总局自2015年7月开展了药物临床试验数据核查,核查中发现部分研究者临床试验意识不强,未充分履行研究者的职责[9]。本研究通过FDA网站,检索2013-2017年FDA发给研究者的警告信,收集违反21 CFR 312.60、21 CFR 812.100条款“研究者总体职责”缺陷项的具体问题,进一步整理与分析,旨在为加强我国临床试验研究者充分履行其职责提供科学建议。
1 资料和方法 1.1 资料本研究资料来自FDA网站(https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/default.htm),检索得到FDA于2013-2017年发出的警告信。
1.2 方法导出警告信的相关信息,内容包括警告信息发布日期(letter issue date)、公司名称(company name)、发布办公室(issuing office)、主题(subject)及终止日期(close out date),保存为Excel格式。使用软件Excel 2007,以subject列的“investigator”为关键词,筛选出发给研究者的警告信。为了保证检索的完整性,核对《美国FDA药物临床试验与非临床研究警告信汇编(2008- 2017)》 [10]目录中“给GCP警告信-给研究者”部分,补充漏检索的警告信。通过阅读每封警告信,筛选包含“21 CFR 312.60”和“21 CFR 812.100”条款的缺陷项。通过Excel 2007软件统计警告信发布日期、研究者姓名、关于“研究者总体职责”缺陷项的具体问题和终止日期,进一步对具体问题进行分类和汇总。
2 结果 2.1 检索结果采用上述方法,从FDA网站检索出3182封警告信。以subject列的“investigator”为关键词,筛选出发给研究者的警告信37封;又通过核对《美国FDA药物临床试验与非临床研究警告信汇编(2008-2017)》目录中“给GCP警告信-给研究者”的部分,发现漏检1封;最终共检索出38封。其中,有35封涉及缺陷项“研究者总体职责”(21 CFR 312.60、21 CFR 812.100)。
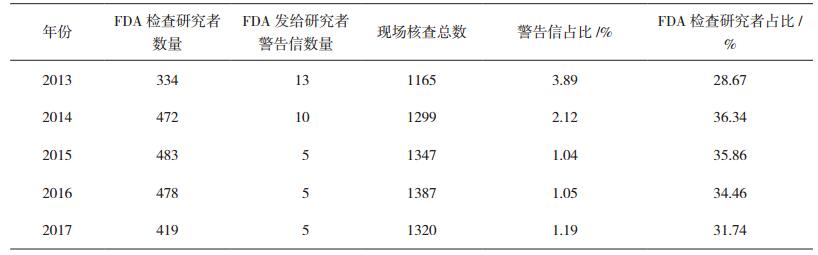
2.2 FDA给研究者警告信的分布情况2013-2017年,FDA共开展现场检查6518次,针对研究者有2168次[11],发出警告信38封,其中针对药物、生物制剂、医疗器械与放射健康临床试验分别是32、2、4封。FDA检查研究者数量占现场检查总数的28.67%~31.74%,比例基本稳定;发给研究者警告信数量占检查研究者数量的1.04%~3.89%,呈逐年下降趋势,见表 1。
|
|
表 1 2013-2017年FDA给研究者警告信分布情况 |
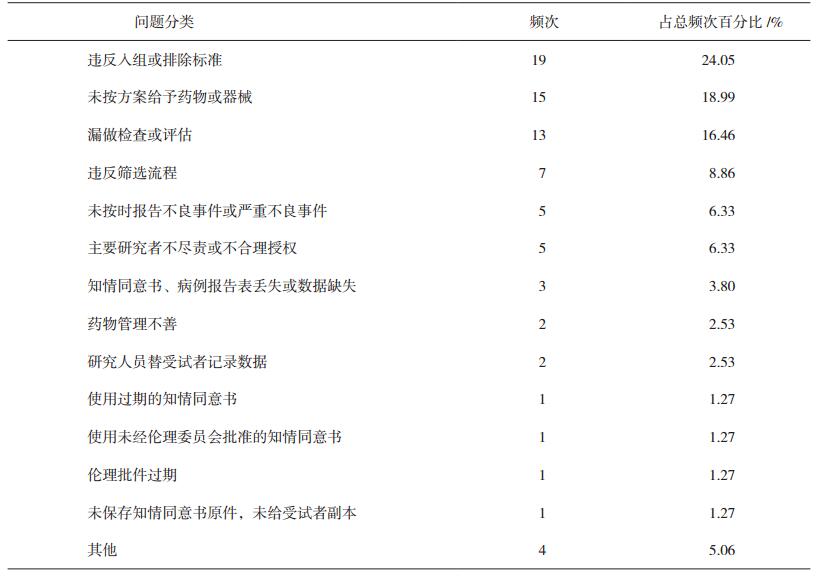
关于“研究者总体职责”缺陷项的具体问题可归纳为14类,具体情况见表 2。
|
|
表 2 “研究者总体职责”缺陷项具体问题的分类情况 |
涉及缺陷项“研究者总体职责”的35封警告信中,有4位研究者及时针对警告信中违规情况做出回复,回复被FDA认可后,FDA网站公布了终止信内容。4封终止信中,仅有发给Michael Ring, MD的终止信[12]描述了研究者采取的纠正措施,例如:对筛选检查早于签署知情同意书的3名受试者重新签署知情同意书;修订标准操作规程(SOP),表明研究者将在任何研究干预之前签署同意书,同时,强化协调研究者在研究中的责任,及时报告安全事件。其余3封警告信均描述“FDA已经根据原警告信评估研究者的纠正措施,认为研究者已经解决了本警告信中的违规行为。在未来的检查和监管活动中,FDA将进一步评估纠正措施的充分性和可持续性……” [13-15]。
3 讨论 3.1 FDA警告信中“研究者总体职责”缺陷项的常见问题分析2013-2017年,FDA发给研究者的警告信中,关于“研究者总体职责”缺陷项的具体问题,发生比例大于5%的问题包括:1)违反入组或排除标准:主要表现为入组前,未完成实验室检查;疾病诊断不符合入组标准;或实验室检查结果远超正常值,符合排除标准。2)未按方案给予药物或器械:主要表现为研究者未按受试者病情的变化,及时调整剂量;或给予错误的剂量。3)漏做检查或评估:主要表现为随访时,受试者漏做实验室检查或其他医学检查;或未填写相应的病情评估。4)违反筛选流程:主要表现为签署知情同意书前,受试者已经完成相应的实验室检查;或在获得实验室检查结果前就完成随机分组。5)未按时报告不良事件(AE)或严重不良事件(SAE):主要表现为研究者未在规定的时限内报告SAE;或针对按SAE管理的AE,研究者未及时报告;或漏报SAE。6)主要研究者不尽责或不合理授权:主要表现为被授权的研究者不具备相应的专业知识,且未经培训。表 2数据显示,上述发生比例大于5%的问题累计总占比超过80%,在“研究者总体职责”缺陷项的问题中占比较大,也警示我国研究者在临床试验实施过程中应严格执行临床试验方案,关注受试者安全和授权合适的研究者。
3.2 我国临床试验研究者职责方面的常见问题对比FDA警告信,我国研究者履行职责方面也存在着类似问题,例如:违反入组或排除标准、知情同意书签署日期晚于筛选日期、故意修改受试者筛选期症状或诊断以满足试验入组标准、药物管理不善、原始数据不一致或丢失、漏报不良事件或严重不良事件等[9, 16]。但与FDA警告信关于研究者职责不同的是,我国临床试验主要研究者通常由医疗机构的业务科室主任或医疗机构领导担任,一般委托协调研究者负责日常工作。因为临床工作繁重,主要研究者缺少对临床试验的日常监督;协调研究者由于资历较浅,很难开展切实有效的监督和协调。另一方面,临床试验数量日益增加,临床研究协调员越来越被广泛使用,部分研究者存在依赖心理,常委托其超出授权范围以外的工作[17]。
3.3 加强我国临床试验研究者职责的建议研究者受申请人委托具体实施临床试验项目,必须保证试验行为符合药物临床试验质量管理规范(GCP)规定,保证试验数据真实、完整、规范及可溯源[18]。为进一步提高我国临床试验质量,保护受试者的权利、安全、福利,有必要采取一定的措施来加强研究者总体职责。结合美国研究者履行职责方面存在的问题,提出几点建议:1)加强信息透明度,完善奖惩制度。目前,临床试验机构仅限于医疗机构,医疗机构应完善相关制度,保障临床试验研究者收入水平[19],同时,针对违规行为给予相应的处罚。我国相关监管部门可根据国情,借鉴FDA经验,颁发相应的法规、文件,将现场检查中发现的研究者相关问题公之于众,研究者在规定时限内提交整改措施,获得认可后,警告信才被终止。上述措施,既能鼓励研究者认真开展临床试验,提高临床试验质量,又能让研究者及时获知临床试验的不规范行为,起到警示作用。2)加强培训,提高研究者意识。加强对主要研究者的培训,及时传达临床试验相关法规、文件,强化临床试验就是临床研究的概念,培养其严谨的临床研究质量意识[20-21]。改变传统的培训方式,采用角色扮演和质控实操的形式,将典型问题做成案例,模拟药物临床试验检查现场,让两组人员分别扮演检查专家和研究者,相互问答,增强研究者的参与度和获得感,从而避免临床试验中的不规范行为。3)授权合格的研究者,加强监督。研究者需经过GCP等法规、本机构管理制度与标准操作规程的培训。为了保证临床试验质量,主要研究者应根据试验方案和涉及的专业,授权负责任的研究者参加临床试验。临床研究协调员应充分发挥协调和监督作用,定期向主要研究者汇报临床试验的进展和问题。
综上所述,研究者在临床试验实施过程中发挥着重要的作用。FDA警告信中关于“研究者总体职责”缺陷项的问题,对我国临床试验的实施起到警示作用。研究者应充分履行其职责,使临床试验得出可靠可信的结果,给我国药品监管部门决策提供科学参考依据,进一步提高我国临床试验整体质量。
(致谢:在撰写论文过程中,吴晓初老师给予了很大的帮助,特此感谢。)
| [1] |
国家药品监督管理局.药物临床试验质量管理规范[Z]. 2003.
|
| [2] |
谢洁琼. 药物临床试验质量控制与质量保证体系探讨[J]. 中国药师, 2015, 18(7): 1191-1194. DOI:10.3969/j.issn.1008-049X.2015.07.040 |
| [3] |
FDA. Electronic Code of Federal Regulations[EB/OL].[2019-01-05]. https://www.ecfr.gov/cgi-bin/ECFR?page=browse. 2018.
|
| [4] |
FDA. FDA WARNING LETTERS: Trends and Perspectives[EB/OL].[2019-01-05]. https://oig.hhs.gov/oei/reports/oei-09-97-00380.pdf. 1999.
|
| [5] |
段然. 浅谈美国FDA警告信的作用[J]. 中国药事, 2013(3): 332-333. DOI:10.3969/j.issn.1002-7777.2013.03.022 |
| [6] |
李小芬. 美国食品和药物管理局针对伦理委员会发出警告信的分析[J]. 中国新药与临床杂志, 2018(11): 619-622. |
| [7] |
FDA. Warning Letters[EB/OL].[2019-01-05]. https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/default.htm. 2018-12-19.
|
| [8] |
FDA. About Warning and Close-Out Letters[EB/OL].[2019-01-05]. https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/ucm278624.htm. 2009.
|
| [9] |
王佳楠, 钱雪, 李见明. 药物临床试验数据核查工作及常见问题分析[J]. 中国新药杂志, 2018, 27(11): 1273-1276. |
| [10] |
国家食品药品监督管理总局食品药品审核查验中心, 上海药品审评核查中心.美国FDA药物临床试验与非临床研究警告信汇编(2008~2017)[M].第1版.北京: 中国医药科技出版社. 2018.
|
| [11] |
FDA. BIMO Inspection Metrics[EB/OL].[2019-01-05]. https://www.fda.gov/ScienceResearch/SpecialTopics/RunningClinicalTrials/ucm261409.htm.
|
| [12] |
FDA. Michael Ring, MD-Close Out Letter[EB/OL].[2019-01-05]. https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/ucm445449.htm. 2015.
|
| [13] |
FDA. Janet K Tillisch, MD-Close Out Letter 7/22/13[EB/O L].[2019-01-05]. https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2013/ucm362058.htm. 2013.
|
| [14] |
FDA. George C. Velmahos, M.D., Ph.D.-Close Out Letter[EB/OL].[2019-01-05]. https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/ucm380581.htm. 2014.
|
| [15] |
FDA. Moussa C. Mansour, MD-Close Out Letter[EB/OL].[2019-01-05]. https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2016/ucm499409.htm. 2016.
|
| [16] |
何高丽, 曾涛, 张炜, 等. 药物临床试验数据核查临床部分常见问题的原因分析及控制措施[J]. 中国新药与临床杂志, 2018, 37(1): 24-28. |
| [17] |
张赟赟, 邢丽娜, 杨帆平, 等. 探讨临床研究协调员的管理作用[J]. 中国临床药理学杂志, 2016, 32(20): 1911-1913. |
| [18] |
国家食品药品监督管理总局.总局关于药物临床试验数据核查有关问题处理意见的公告2017年第63号[S]. 2017.
|
| [19] |
中共中央办公厅, 国务院办公厅.关于深化审评审批制度改革鼓励药品医疗器械创新的意见[S]. 2017.
|
| [20] |
汪朝晖, 杨忠奇, 杜彦萍, 等. 谈药物临床试验中研究者依从性的管理[J]. 江西中医学院学报, 2009, 21(1): 20-22. DOI:10.3969/j.issn.1005-9431.2009.01.009 |
| [21] |
张琳, 曾涛, 张炜, 等. 美国食品和药物管理局针对临床试验研究者发出警告信的分析[J]. 中国新药与临床杂志, 2017(8): 451-454. |