 2019, Vol. 33
2019, Vol. 33 
2. 中国科学院兰州化学物理研究所, 兰州 730000
2. Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou 730000, China
近年来, 随着组合化学和高通量筛选技术在新药研发中的广泛应用, 越来越多的新药面临溶解度低、溶出速率慢、口服生物利用度差的问题, 提高难溶性药物的溶出性能和口服生物利用度已经成为药剂学研究的热点[1]。许多研究表明将这些难溶性药物制成无定形是增加其溶出速率和口服生物利用度的有效方法之一[2]。相对于晶型药物, 无定形药物具有较大的表面自由能, 在溶出过程中, 无需克服晶格能的限制, 因此, 具有较高的溶解度和溶出速率。然而, 由于无定形药物处于热力学高能态, 在制备、贮存和溶出的过程中容易发生重结晶, 并可能导致溶出性能和体内药效的改变, 这种物理不稳定性严重限制了无定形药物的实际使用[3-4]。
目前, 提高无定形药物物理稳定性的方法主要包括将无定形药物包载在聚合物或介孔硅材料中制备成固体分散体或介孔硅给药系统。然而, 固体分散体和介孔硅给药系统的制备需要加入大量的水溶性聚合物和介孔硅材料, 载药量有限, 制备工艺仍不成熟, 工业放大生产困难[5]。这些缺点限制了固体分散体和介孔硅给药系统的商业化应用。近年来, 共无定形药物作为一种新颖的药物固体形态, 能够提高无定形药物的物理稳定性, 显著改善难溶性药物的溶解度和溶出速率, 具有制备工艺简单、载药量大的优点, 弥补了上述两种方法的不足[6]。此外, 将两种具有协同药理作用的药物制备成无定形给药系统, 对解决仿制药物和复方药物开发的专利挑战具有重要意义。因此, 无定型药物在学术和制药工业领域受到广泛关注[7-8]。
本文就共无定形药物的载体材料选择、制备工艺、物理稳定机制、体外溶出性能和体内吸收情况, 以及未来发展前景进行了综述。
1 共无定形药物概述固体分散体(Solid Dispersions, SD)是指药物以分子、无定形或微晶状态高度均匀分散在固体载体材料中的给药系统。自1961年固体分散技术首次用于提高难溶性药物的溶出速率和口服生物利用度以来, 诸多研究者对固体分散体进行了广泛深入的研究, 发表了大量论文, 进一步证明将难溶性药物制成无定形固体分散体是改善其溶解度和溶出速率的最具应用潜力的方法之一[9-10]。按照药物在载体材料中的物理分散状态, 固体分散体可以分为低共熔混合物、固态溶液、玻璃溶液和玻璃混悬液, 详见图 1[7-8]。按照载体材料的种类, 固体分散体可以分为大分子固体分散体(聚合物固体分散体和介孔硅固体分散体)和小分子固体分散体。当固体分散体中药物以分子或无定形存在, 且载体材料为其他小分子药物或者小分子药用辅料时, 此种固体分散体即为共无定形药物[7-8]。因此, 共无定形药物仍属于固体分散体研究的一个分支[11]。尽管此前曾有零散的小分子固体分散体研究, 但并没有引起足够的重视, 近年来, 随着传统大分子固体分散体缺点的暴露, 共无定形药物研究日益得到重视[12-13]。2009年, Chieng等[6]采用球磨法制备得到了吲哚美辛-盐酸雷尼替丁无定形混合物, 提高了无定形药物的物理稳定性, 改善了药物的溶解度和口服生物利用度, 并且盐酸雷尼替丁可以降低吲哚美辛的胃肠道副作用。同时, 为了进一步区分于聚合物固体分散体, 作者首次提出了共无定形药物(Coamorphous)这一概念。此后有关共无定形药物的研究论文日益增多, 并成为了药剂学前沿研究的热点之一[14-15]。目前, 国内外关于共无定形药物尚无统一定义, 本文综合相关文献资料, 将无定形药物的定义概括为活性药物成分和其他药物或辅料等小分子固体组分, 通过氢键、离子键等非共价键作用或者简单的物理混合后, 形成的二元单相无定形固体分散体给药系统。

|
图 1 固体分散体的分类 |
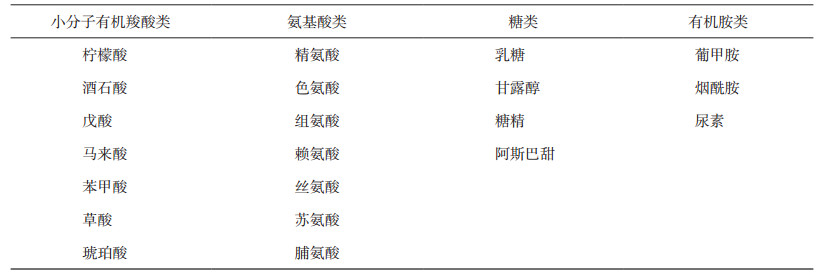
根据共无定形药物配体组分的不同, 可以将其分为两类:一类为药物-药物共无定形。通过将具有协同药理作用或者联合用药的两种药物结合制备成共无定形药物, 提高无定形物理稳定性的同时, 提高药物疗效、降低药物的毒副作用。另一类为药物-小分子药用辅料共无定形。常用于制备共无定形药物的药用辅料主要包括安全无毒的小分子有机羧酸类、氨基酸类、糖类和有机胺类, 已报道的常见小分子配体[16-20]见表 1。目前, 有关共无定形药物配体的选择尚无共同规律, 其主要指导原则是依据药物的化学结构和理化性质(氢键供体和受体、酸碱性、解离基团、芳香环等), 选择与之具有良好固态相容性的药物或者药用辅料, 通过处方前的大规模筛选得到。其筛选的流程和方法类似共晶筛选[15]。目前, 已报道的配体筛选指导方法包括溶解度参数法[21-22]、Flory-Huggsins方程作用参数法[23-24], 以及计算机模拟辅助筛选法[25-27]。
|
|
表 1 制备共无定形药物的常见配体 |
在已报道的共无定形药物的制备工艺中, 根据机理的不同, 这些工艺可以分为热力学方法和动力学方法(见图 2)。其中热力学方法主要是将药物熔融后淬冷, 使其迅速玻璃化, 或者将药物溶于有机溶剂中, 再快速去除溶剂后得到共无定形药物, 包括旋转蒸发法、熔融淬冷法、冷冻干燥法和喷雾干燥法等。动力学方法是利用碾磨过程中的冲击、磨擦和剪切作用不断破坏药物的晶体结构, 使其转变为无定形态, 包括球磨法、研磨法、溶剂辅助研磨法和热熔挤出法等。其中喷雾干燥法和热熔挤出法克服了共无定形药物实验室制备的缺陷, 有望实现共无定形药物工业化生产。本节对热力学和动力学的各代表方法逐一进行了介绍, 并详述了喷雾干燥法和热熔挤出法。

|
图 2 共无定形药物的制备方法 |
该法是通过将均匀混合的样品在隔绝空气或者氮气保护的条件下, 加热至完全熔融后, 置于液氮中淬冷, 干燥后即得共无定形药物。该法将药物熔融后快速冷却, 使药物分子来不及重新排列, 从而抑制药物晶核的生成和晶体的生长。该方法简便, 无需有机溶剂, 适用于对热稳定、不易分解的药物。Lobmann等[28-29]利用熔融淬冷法制备了不同摩尔比的吲哚美辛-萘普生共无定形药物, 并研究了药物分子间的相互作用对共无定形体系药物溶出和药物重结晶的影响。研究结果发现, 相对于单个药物, 共无定形药物的物理稳定性显著提高。等摩尔比的吲哚美辛-萘普生共无定形药物显示最佳的物理稳定性, 并且在溶出介质中能够以相同速度同时释放。傅里叶转换红外光谱法和Gordon-Taylor方程模拟结果显示, 吲哚美辛和萘普生通过其化学结构上的羧酸基团的氢键作用形成了异质二聚体, 从而改变了药物分子的重结晶和溶解行为。
3.2 溶剂挥发法该法是通过将样品溶解于合适的有机溶剂后, 通过减压加热干燥或者喷雾干燥, 快速挥干有机溶剂得到共无定形药物。目前, 常用的有机溶剂包括甲醇、乙醇、乙腈、丙酮、二氯甲烷等。该方法适用于热敏感药物共无定形的实验室制备, 缺点是不适用于工业化生产, 制备得到的无定形药物的粉体流动性和可压性较差, 不利于压片和填充胶囊。Qian等[30]利用溶剂挥发法制备了盐酸鲁拉西酮-糖精共无定形药物, 共无定形药物显著提高了盐酸鲁拉西酮的溶解度并且物理稳定性得到改善, 共无定形药物在25℃/60%湿度条件下贮存60天后仍然保持无定形态, 而无定形盐酸鲁拉西酮在相同的条件下2天内就出现了重结晶。Dengale等[31]利用溶剂挥发法制备了槲皮苷和利托那韦共无定形药物, 共无定形药物利托那韦的饱和溶解度是结晶药物的5倍, 其中1︰2和2︰1摩尔比的无定形药物在40 ℃干燥条件下保持物理稳定达90天。
3.3 球磨法该法通过将样品置于球磨机中, 利用高速冲击、磨擦和剪切等机械作用对样品进行充分混合, 并破坏样品的晶体结构, 使其转变为共无定形态。研磨过程中, 高速冲击、磨擦和剪切等机械作用产生大量的热能, 使样品温度升高, 可能会导致热敏感药物的降解, 促进无定形药物的重结晶。因此, 在研磨过程中采取降温措施, 进行低温研磨, 或者在样品中加入少量溶剂进行研磨都可以加快共无定形药物转化效率。Allesø等[32]采用低温球磨法分别制备了1︰2、1︰1和2︰1摩尔比的萘普生-西咪替丁共无定形药物, 其中1︰1摩尔比的样品物理稳定性最佳, 在40 ℃干燥条件下保持稳定长达186天, 其特性溶解速率分别是萘普生和西咪替丁特性溶解速率的4倍和2倍。Jensen等[33-34]利用球磨法分别加入各种色氨酸和精氨酸, 制备了萘普生和吲哚美辛共无定形药物, 研究结果发现, 在研磨过程中药物能够和氨基酸逐渐互溶, 从而形成无定形盐, 显著改善了药物的溶解度和溶出速率。
3.4 喷雾干燥法相对于传统的实验室规模的溶剂挥发法, 喷雾干燥法是工业化制备无定形药物的成熟工艺。目前已有一系列研究报道利用喷雾干燥法制备共无定形药物[35-37]。喷雾干燥法制备无定形药物的基本过程是首先将药物和配体溶于水或混合溶剂中, 溶液通过离心或机械加压雾化后喷入干燥室, 与热气流接触后溶剂迅速挥发, 从而得到干燥产品。该方法生产效率高, 工艺简单, 产品为粉末或颗粒状, 无需进一步粉碎, 可直接和辅料混合后压片或填充胶囊。Jensen等[38]利用喷雾干燥法制备了吲哚美辛-精氨酸、吲哚美辛-组氨酸、吲哚美辛-赖氨酸共无定形药物, 研究发现三种共无定形药物在40℃干燥条件下保持稳定长达10个月以上, 相对于吲哚美辛-组氨酸、吲哚美辛-赖氨酸共无定形药物, 吲哚美辛-精氨酸共无定形药物显著改善了药物的溶解度, 而吲哚美辛-组氨酸、吲哚美辛-赖氨酸共无定形药物由于在溶出过程中吲哚美辛发生重结晶, 其溶出没有显著改善。Jaya等[39]进一步研究了喷雾干燥溶剂组分对吲哚美辛-精氨酸、吲哚美辛-组氨酸、吲哚美辛-赖氨酸共无定形药物理化性质的影响, 研究结果表明不管溶剂是丙酮还是水-乙醇混合物, 吲哚美辛均能和上述三种氨基酸形成稳定的共无定形, 然而溶剂的配比和种类对共无定形的生产具有重要影响。
3.5 热熔挤出法热熔挤出法是制备共无定形药物较为新颖的方法, 该方法的特点是无需有机溶剂, 可以连续化生产, 制备效率高, 但要求药物和配体具有较高的热稳定性。目前, 热熔挤出法是制备固体分散体的常见方法, 并已有成熟的商品化产品上市, 而热熔挤出法在共无定形药物的制备应用仅有零星的报道。热熔挤出法制备无定形药物的基本过程是将药物、配体以及合适比例的聚合物混合均匀后, 加入到螺杆挤出机中, 物料通过机筒的加热熔融, 以及螺杆挤压、剪切和搅拌等作用达到分子水平的混合后从模口端挤出成型。Arnfast等[40]利用热熔挤出法制备了吲哚美辛-西咪替丁无定形药物, 研究结果发现处方中加入5%聚氧化乙烯可显著降低熔体的粘度, 避免了共无定形药物在贮存过程中的相分离。Lenz等[41]同样利用溶剂辅助热熔挤出法制备了吲哚美辛-精氨酸共无定形药物。他们首先将精氨酸溶于水溶液中, 然后注入到热熔挤出机中与药物辅料进行混合挤出, 保证药物和精氨酸能够充分混合, 挤出物粉碎后放入真空干燥箱中除去剩余的水分。通过这种工艺, 在无需加入聚合物的条件下, 制得了吲哚美辛-精氨酸无定形药物, 处方中加入适量的聚维酮显著提高了共无定形药物的溶解速度。
3.6 小结目前, 共无定形药物制备技术还处于实验室基础研究阶段, 大多数研究主要是通过熔融淬冷法、溶剂挥发法和球磨法等实验室规模进行制备来加以研究。这些方法相对简便、快速, 适用于小样本的筛选研究, 但每一种方法均有一定的缺陷。因此, 制备共无定形药物时, 需要根据药物和共无定形配体的理化性质, 如药物的熔点、热稳定性、药物在溶剂中的溶解度等参数合理加以选择。随着共无定形药物研究的成熟, 共无定形药物的大规模工业化生产方法已经有所报道, 这些方法包括喷雾干燥法、热熔挤出法和冷冻干燥法等。此外, 随着个体化治疗的发展, 已有研究报道利用3D打印技术制备吲哚美辛-精氨酸共无定形药物, 从而有望实现更加灵活和针对性的治疗[42]。
4 共无定形药物物理稳定性机制 4.1 固溶度共无定形体系是由两种或多种小分子组分组成的一种单相无定形混合物。对于单相的混合物体系, 体系中的各组分需要在无定形状态时可以完全相溶[11]。药物在载体材料中的溶解度越大, 则体系的稳定性越好。反之, 如果药物在载体材料中过饱和, 则共无定形体系具有较高的化学势能, 药物分子易于重结晶。溶解度参数可用于检查共无定形混合物中热不稳定化合物的相容性[21]。两种互溶组分形成均匀单相共无定形混合物的另一个指标是可以观察到单一的玻璃化转变温度(Tg)。相反地, 不互溶或部分互溶的组分会形成两相无定形混合物, 从而产生两个Tg[15]。
相应的, 对于聚合物基玻璃溶液, 药物在无定形聚合物中的热力学溶解度被认为是稳定性的主要原因之一, 但是许多药物在聚合物载体中的溶解度都很有限[43]。当药物在聚合物中过饱和时, 会出现药和(或)聚合物/辅料相分离, 然后快速成核, 晶体生长。同样, 制备物理稳定的共无定形药物要求组分之间能够互溶, 而部分互溶或不互溶的均匀混合的共无定形混合物会迅速出现相分离和结晶。Jensen等[33]对吲哚美辛-色氨酸和呋塞米-色氨酸共无定形药物的形成机制进行了研究, 结果表明在球磨过程中药物的X射线粉末衍射峰逐渐消失, 并最终得到了共无定形药物。结合差示扫描量热法的结果, 作者推测吲哚美辛-色氨酸共无定形的形成机制是色氨酸溶于吲哚美辛, 而呋塞米-色氨酸共无定形药物是呋塞米逐渐溶于色氨酸中。综上所述, 制备稳定的共无定形药物要求药物和载体材料之间具有良好的相容性和固溶度。
4.2 玻璃转化温度玻璃化转变是无定形相从冻结状态到解冻状态的一种弛豫现象。当温度达到Tg时, 物质从固态变为粘弹态, 分子运动加剧。由于分子迁移率增加, 物质在过冷液相态时结晶的速度远远大于无定形玻璃态。因此, 为了保持玻璃态物质的稳定性, 建议一般将无定形药物放置在低于Tg 50℃的温度下贮存[44]。共无定形药物的玻璃转化温度反映了分子的迁移率, 可以作为物理稳定性评价的重要指标。一般来说, 玻璃化转变温度越高, 分子迁移率越低, 物理稳定性越好。大量的研究结果表明共无定形药物的玻璃化转变温度大于单一组分的玻璃化转变温度, 这也是共无定形体系物理稳定性提高的重要原因之一[45]。Lobmann等[18, 46]利用球磨法制备了卡马西平、吲哚美辛与各种氨基酸的共无定形药物, 研究结果表明药物和氨基酸形成共无定形药物后, 相对于单一的药物, 共无定形体系的玻璃化转变温度显著增加。共无定形药物在40℃干燥条件下保持稳定长达6个月, 而单一的无定形药物在同样的条件下仅7天就出现了重结晶。红外光谱数据结果表明吲哚美辛和氨基酸通过离子键产生了成盐反应, 而卡马西平和氨基酸主要通过氢键和π-π相互作用从而抑制了药物的重结晶。Kissi等[45]对玻璃化转变温度下的共无定形药物的α和β分子弛豫现象进行了详细研究, 研究结果表明α分子弛豫温度对配比不均衡的无定形药物体系的物理稳定性没有指导意义, 而β分子弛豫温度能够进一步反映体系的物理稳定性。综上所述, 玻璃化转变温度是预测共无定形药物物理稳定性的重要指标, 同时, 药物和其他配体之间的相互作用对体系的物理稳定性也具有决定性的影响。
4.3 分子间的相互作用许多研究表明共无定形体系比单一的无定形药物的物理稳定性要好, 其原因除了玻璃化转变温度的改变外, 无定形混合物中组分分子间的相互作用是物理稳定性提升的重要原因。这些分子间的相互作用包括氢键作用、离子键作用或π-π相互作用等[12]。由于药物和其他小分子的相互作用, 从而降低了分子迁移率, 抑制了药物成核和晶体生长。Fung等[47]利用喷雾干燥法分别制备了草酸-酮康唑、酒石酸-酮康唑、柠檬酸-酮康唑、琥珀酸-酮康唑等摩尔比的共无定形药物, 并利用傅里叶转化红外光谱、介电光谱和固态核磁共振对药物和其他小分子有机酸之间的相互作用进行了详细研究。研究结果表明药物和其他分子间的相互作用直接决定了药物的分子迁移率和物理稳定性, 酮康唑与草酸、酒石酸、柠檬酸为离子键相互作用, 而酮康唑和琥珀酸以氢键作用为主, 分子作用越强, 则体系的物理稳定性越好。同时, 他们也发现分子迁移率并不能直接预测体系的物理稳定性, 配体的化学结构和空间位阻对物理稳定性也有一定的影响。
4.4 均匀混合值得一提的是在某些共无定形药物体系中, 现有的表征方法没有探测到药物和配体材料之间存在氢键、离子键或π-π等相互作用, 其物理稳定性的提高可能是药物和配体材料分子水平均匀混合后产生的空间位阻所致。Knapik等[48]利用熔融淬冷法制备了依泽替米贝-吲达帕胺共无定形药物, 并从分子动力学角度研究了其物理稳定性机制, 实验结果表明不同摩尔配比的共无定形药物的玻璃化转变温度随吲达帕胺比例的增加而增加, 其不同配比的共无定形药物的玻璃化转变温度结果和Gordon-Taylor方程的预测结果一致, 共无定形药物在25℃/25%湿度条件能够保持物理稳定72天, 差示扫描量热法、傅里叶转化红外光谱和介电谱研究结果表明药物之间不存在相互作用, 其物理稳定性提高的主要原因是吲达帕胺的反增塑作用。Dengale等[49]报道利用溶剂蒸发法制备了利托那韦-吲哚美辛共无定形药物, 其不同配比的共无定形药物的玻璃化转变温度结果和Gordon-Taylor方程的预测结果没有显著差异, 同时, 傅里叶转换红外光谱结果显示利托那韦和吲哚美辛之间不存在氢键或离子键作用, 相对于对应的结晶药物, 共无定形药物的溶解度提高了3倍。
4.5 小结综上所述, 对大多数共无定形体系, 其物理稳定提升机制可能是上述几种因素综合作用的结果。其中, 良好的固溶度和均匀混合是共无定形药物稳定的前提, 药物和配体之间的相互作用是共无定形药物稳定的充分不必要条件, 玻璃化转变温度是预测和评估共无定形体系物理稳定性的重要指标。不同因素之间相互影响, 共同维系无定形体系的物理稳定性。
5 溶解特性由于无定形态具有较高的内能, 溶解时无需克服药物的晶格能, 因此, 和药物的结晶态相比, 无定形态具有较高的溶解度和溶解速率。大量的研究结果表明共无定形体系的溶解行为不仅比它们的晶态提升了, 而且优于单一的无定形组分[11]。药物和配体之间的相互作用不仅可以防止溶剂介导的药物重结晶, 而且可以加速药物的溶解, 实现药物的同步释放和维持药物的过饱和状态。
Allesø等[32]研究了萘普生-西咪替丁共无定形的溶解速率, 实验结果表明共无定形的溶解速率高于晶体药物和无定形西咪替丁。无定形西咪替丁的溶解速率和西咪替丁晶体的相同, 说明无定形西咪替丁在接触溶质后迅速重结晶, 导致了无定形和晶体药物的溶出速率没有差别。但是当萘普生与西咪替丁形成共无定形后, 两种药物可以实现同步释放, 同时萘普生和西咪替丁的溶解速率分别提高了4倍和2倍, 而且在溶出过程中没有发现药物重结晶的现象, 作者认为共无定形药物分子之间的相互作用阻止了药物在溶出过程中的重结晶。Qian等[50]在盐酸鲁拉西酮-糖精共无定形药物体系中发现了相似的试验结果, 无定形盐酸鲁拉西酮在溶解过程中可以观察到溶剂介导的快速重结晶, 导致无定形态与结晶态的溶解速率相比没有显著差异。与此相反, 盐酸鲁拉西酮-糖精共无定形药物在溶解试验的整个阶段没有出现药物的重结晶现象, 溶出速率比结晶态提高了5.6倍。傅里叶转化红外光谱结果表明盐酸鲁拉西酮和糖精分子之间存在的较强电荷辅助氢键作用抑制了溶解过程中溶剂引起的药物重结晶。分子间的相互作用在药物的溶出过程中发挥了重要作用[29, 38]。
除了溶解速率增加, 共无定形药物组分呈现短程有序分子序列, 即通过分子之间相互作用形成异质二聚体, 实现了药物同步释放[24, 32]。萘普生-西咪替丁共无定形药物由于形成了异质二聚体, 溶出过程中药物相互关联, 导致同步释放。同样的原理, 萘普生-吲哚美辛共无定形药物也出现了类似的现象[29]。无定形药物的同步释放和两种药物分子间的相互作用密切相关。
共无定形药物的同步释放通常是因为组分间较强的相互作用导致的, 因此, 可以通过调节共无定形药物配体的溶解度或溶解速率来调整难溶性药物的溶解速率。当两种组分间存在强分子间相互作用时, 易溶性的配体可以辅助难水溶性药物的溶解。一些研究表明吲哚美辛-氨基酸共无定形体系的溶解速率取决于氨基酸配体的溶解性[45-46]。Jensen等[32, 36]通过在处方中加入易溶性的配体提高了无定形萘普生的溶解速度, 该处方中加入具有和萘普生强相互作用的精氨酸, 增强无定形药物的物理稳定性。此外, 处方中加入易溶的脯氨酸改善了体系的溶解性。相对于萘普生/脯氨酸或萘普生/精氨酸二元混合物, 三元无定形混合物的溶解速率显著增加。但是, 随后的研究发现, 多组分的共无定形药物只能在一定程度上增强药物的溶出速率, 当药物的溶出速率过快或组分间相互作用不够强时, 溶出介质中的过饱和药物则会沉淀析出[34, 38]。
共无定形药物提高难溶性药物生物利用度的另外一种可能机制是维持了药物过饱和状态, 延长了药物的吸收时间。目前, 有关于共无定形药物在模拟肠液中的过饱和溶出行为的研究较少, 有待于进一步探索[11]。Paluch等[51]分别利用喷雾干燥法和球磨法制备了2︰1和1︰1的环丙沙星琥珀酸共无定形药物, 溶出试验结果表明2︰1的环丙沙星琥珀酸共无定形盐优于1︰1的无定形盐, 共无定形药物在溶出介质中保持较长时间的饱和状态, 无定形环丙沙星的浓度可以达到58.8 mg·mL-1, 并维持1个小时以上, 而环丙沙星结晶药物的浓度小于0.1mg·mL-1。Heikinnen等[52]研究了不同的氨基酸药物共无定形制剂在磷酸缓冲液和胃肠(禁食和非禁食条件)模拟液的溶解情况。和单独的无定形药物或晶形药物相比, 所有的共无定形药物(辛伐他汀-赖氨酸、丝氨酸-苏氨酸、格列本脲-苏氨酸、格列本脲-丝氨酸-苏氨酸)的溶解速率都显著增加, 药物过饱和状态维持时间显著延长。与此类似, Ojarinta等[53]研究了吲哚美辛氨基酸共无定形药物在相同溶出介质中的药物释放行为, 研究结果表明药物的过饱和状态取决于溶出介质的种类以及药物和配体之间的相互作用。Petry等[54]进一步研究了包衣对吲哚美辛-精氨酸共无定形药物的物理稳定性、药物释放和生物利用度的影响, 研究结果表明包衣过程并没有诱导药物在贮存过程中的重结晶, 反而抑制了药物在溶出过程中的重结晶, 并在溶出介质中产生了更高的过饱和。值得一提的是并不是所有的共无定形药物都能够保持药物的过饱和状态, 促进药物的透膜吸收。Trasi和Taylor[55]研究了利托那韦、洛匹那韦和紫杉醇在生物模拟液中的过饱和透膜转运, 结果表明第二种药物的加入降低了药物的跨膜转运并促进了药物的相分离。
6 体内性能尽管共无定形药物能够显著改善难溶性药物的体外溶出效果, 但其潜力和优势需要通过改善体内生物利用度来反映。然而, 目前关于共无定形药物的体内研究仍比较缺乏, 大部分共无定形药物的体内研究报道显示, 相对于结晶药物, 共无定形药物的生物利用度显著提高, 但是, 单一的无定形药物或者无定形药物混合物没有作为对照组进行分析, 因此, 生物利用度的提高有可能不是药物共无定形化产生的结果[31, 56]。其中Wang等[57]比较了单一剂量下共无定形药物柠檬酸-氯雷他定和氯雷他定结晶药物的大鼠口服生物利用度差异, 研究结果表明共无定形药物柠檬酸-氯雷他定的生物利用度是结晶药物的2.5倍, 最高血药浓度是结晶药物的2.6倍。Shayanfar等[58]同样发现烟酰胺-阿托伐他汀钙共无定形的生物利用度是结晶阿托伐他汀钙的两倍。如上所述, 以上研究并没有把单独的无定形药物作为对照组, 因此, 难以判断共无定形在改善药物体内性能所起的作用。Moinuddin等[59]的研究结果则进一步表明共无定形药物能够充分发挥药物和配体的协同作用, 从而提高药物的生物利用度。他们利用冷冻研磨法制备了氢氯噻嗪-阿替洛尔共无定形药物, 体内药代动力学结果显示氢氯噻嗪共无定形药物的体内生物利用度>氢氯噻嗪物理混合物>无定形氢氯噻嗪>结晶氢氯噻嗪。共无定形能够提高氢氯噻嗪生物利用度的主要原因是亲水性的阿替洛尔能够抑制溶出过程中氢氯噻嗪的重结晶。
7 结语与展望共无定形处方可以改善水难溶性药物的溶解度, 提高药物的口服生物利用度, 是一种应用前景广阔的制剂技术。目前, 共无定形药物研究是药物制剂领域研究的前沿热点, 相关的文献发表和专利申请逐年增加, 但是, 作为一种崭新的平台技术, 许多方面仍需要进一步的研究。
首先, 共无定形药物的物理稳定机制仍然不明。温度、湿度等外部贮存条件对共无定形药物的物理稳定性影响需要进一步探索。尽管相对于亲水性聚合物固体分散体, 共无定形药物的吸湿性显著降低, 但水分作为一种良好的增塑剂, 其对共无定形药物的物理稳定性影响不容小觑。目前, 共无定形药物的大多数物理稳定性研究是在干燥条件下进行的, 水分对于共无定形体系的影响需要进行广泛深入的研究。
其次, 在制备药物-配体共无定形药物时, 如何合理选择配体仍然缺乏有效的理论指导。目前, 共无定形配体的选择主要基于随机大规模的处方筛选, 需要投入大量的人力和物力, 效率较低, 如何根据药物的化学结构和性质合理地筛选配体是共无定形药物下一阶段研究的重点。另外, 对于药物-药物共无定形处方, 还需要根据药物的药理特性和给药剂量进行处方设计, 难度和挑战更大。目前, 共无定形药物的体外溶出机制和体外-体内相关性研究仍然缺乏, 需要进一步深入研究。
最后, 目前已有报道利用喷雾干燥法、热熔挤出法等规模化生产技术用于共无定形药物的制备, 但是这些工业化方法仍然不成熟, 工艺过程和参数对共无定形药物性能的影响缺乏研究。共无定形药物作为制剂中间体, 需要进一步做成胶囊或片剂等剂型, 但是目前对于共无定形制剂的加工性能的了解还很少。无定形材料通常会出现粉体流动性差、可压性差、粘冲等制剂问题, 需要逐一解决。
尽管存在许多挑战, 共无定形药物仍然是一种非常有潜力和前景的技术。随着研究的深入, 共无定形制剂技术为解决难溶性药物的口服生物利用度问题提供了一种新的选择和有力工具, 并有望在未来实现产品的商业化生产和销售。
| [1] |
Williams Ⅲ R O, Watts A B, Miller D A. Formulating Poorly Water Soluble Drugs[M]. New York: Springer Science & Business Media, 2011.
|
| [2] |
Yu L. Amorphous Pharmaceutical Solids:Preparation, Characterization and Stabilization[J]. Advanced Drug Delivery Reviews, 2001, 48(1): 27-42. |
| [3] |
Lu M, Guo Z F, Li Y C, et al. Application of Hot Melt Extrusion for Poorly Water-soluble Drugs:Limitations, Advances and Future Prospects[J]. Current Pharmaceutical Design, 2014, 20(3): 369-387. DOI:10.2174/13816128113199990402 |
| [4] |
Vasconcelos T, Marques S, Das N J, et al. Amorphous Solid Dispersions:Rational Selection of a Manufacturing Process[J]. Advanced Drug Delivery Reviews, 2016, 100: 85-101. DOI:10.1016/j.addr.2016.01.012 |
| [5] |
Qi S, Craig D. Recent Developments in Micro-and Nanofabrication Techniques for the Preparation of Amorphous Pharmaceutical Dosage Forms[J]. Advanced Drug Delivery Reviews, 2016, 100: 67-84. DOI:10.1016/j.addr.2016.01.003 |
| [6] |
Chieng N, Aaltonen J, Saville D, et al. Physical Characterization and Stability of Amorphous Indomethacin and Ranitidine Hydrochloride Binary Systems Prepared by Mechanical Activation[J]. European Journal of Pharmaceutics and Biopharmaceutics, 2009, 71(1): 47-54. DOI:10.1016/j.ejpb.2008.06.022 |
| [7] |
Dengale S J, Grohganz H, Rades T, et al. Recent Advances in Co-amorphous Drug Formulations[J]. Advanced Drug Delivery Reviews, 2016, 100: 116-125. DOI:10.1016/j.addr.2015.12.009 |
| [8] |
Chavan R B, Thipparaboina R, Kumar D, et al. Coamorphous Systems:a Product Development Perspective[J]. International Journal of Pharmaceutics, 2016, 515(1-2): 403-415. DOI:10.1016/j.ijpharm.2016.10.043 |
| [9] |
Serajuddin A T. Solid Dispersion of Poorly Water-soluble Drugs:Early Promises, Subsequent Problems, and Recent Breakthroughs[J]. J Pharm Sci, 1999, 88(10): 1058-1066. DOI:10.1021/js980403l |
| [10] |
Jermain S V, Brough C, Williams R O. Amorphous Solid Dispersions and Nanocrystal Technologies for Poorly Water-soluble Drug Delivery-An Update[J]. International Journal of Pharmaceutics, 2017, 532(1): 379-392. |
| [11] |
Laitinen R, Lobmann K, Grohganz H, et al. Supersaturating Drug Delivery Systems:The Potential of Co-amorphous Drug Formulations[J]. International Journal of Pharmaceutics, 2017, 532(1): 1-12. DOI:10.1016/j.ijpharm.2017.08.123 |
| [12] |
Korhonen O, Pajula K, Laitinen R. Rational Excipient Selection for Co-amorphous Formulations[J]. Expert Opinion on Drug Delivery, 2017, 14(4): 551-569. DOI:10.1080/17425247.2016.1198770 |
| [13] |
Kavanagh O N, Albadarin A B, Croker D M, et al. Maximising Success in Multidrug Formulation Development:A Review[J]. Journal of Controlled Release, 2018, 283: 1-19. DOI:10.1016/j.jconrel.2018.05.024 |
| [14] |
Grohganz H, Priemel P A, Lobmann K, et al. Refining Stability and Dissolution Rate of Amorphous Drug Formulations[J]. Expert Opinion on Drug Delivery, 2014, 11(6): 977-989. DOI:10.1517/17425247.2014.911728 |
| [15] |
Newman A, Reutzel-Edens S M, Zografi G. Coamorphous Active Pharmaceutical Ingredient-Small Molecule Mixtures:Considerations in the Choice of Coformers for Enhancing Dissolution and Oral Bioavailability[J]. J Pharm Sci, 2018, 107(1): 5-17. DOI:10.1016/j.xphs.2017.09.024 |
| [16] |
Lobmann K, Jensen K T, Laitinen R, et al. Stabilized Amorphous Solid Dispersions with Small Molecule Excipients:Amorphous Solid Dispersions[M]. New York: Springer International Publishing, 2014: 613-636.
|
| [17] |
Hoppu P, Jouppila K, Rantanen J, et al. Characterization of Blends of Paracetamol and Citric Acid[J]. Journal of Pharmacy and Pharmacology, 2007, 59: 373-381. DOI:10.1211/jpp.59.3.0006 |
| [18] |
Lobmann K, Grohganz H, Laitinen R, et al. Amino Acids as Co-amorphous Stabilizers for Poorly Water Soluble Drugs-Part 1:Preparation, Stability and Dissolution Enhancement[J]. European Journal of Pharmaceutics and Biopharmaceutics, 2013, 85(3): 873-881. DOI:10.1016/j.ejpb.2013.03.014 |
| [19] |
Descamps M, Willart J F, Dudognon E, et al. Transformation of Pharmaceutical Compounds upon Milling and Comilling:the Role of Tg[J]. Journal of Pharmaceutical Sciences, 2007, 96: 1398-1407. DOI:10.1002/jps.20939 |
| [20] |
Erizal Z, Fitri R, Fithriani A, et al. Preparation and Characterization of Binary Mixture of Efavirenz and Nicotinamide[J]. Oriental Journal of Chemistry, 2015, 31(4): 2271-2276. DOI:10.13005/ojc |
| [21] |
Alhalaweh A, Alzghoul A, Kaialy W. Data Mining of Solubility Parameters for Computational Prediction of Drug-excipient Miscibility[J]. Drug Development and Industrial Pharmacy, 2014, 40(7): 904-909. DOI:10.3109/03639045.2013.789906 |
| [22] |
Kitak T, Dumičić A, Planinšek O, et al. Determination of Solubility Parameters of Ibuprofen and Ibuprofen Lysinate[J]. Molecules, 2015, 20(12): 21594-21568. |
| [23] |
Pajula K, Taskinen M, Lehto V-P, et al. Predicting the Formation and Stability of Amorphous Small Molecule Binary Mixtures from Computationally Determined FloryHuggins Interaction Parameter and Phase Diagram[J]. Molecular Pharmaceutics, 2010, 7(3): 795-804. DOI:10.1021/mp900304p |
| [24] |
Loebmann K, Strachan C, Grohganz H, et al. Co-amorphous Simvastatin and Glipizide Combinations Show Improved Physical Stability Without Evidence of Intermolecular Interactions[J]. European Journal of Pharmaceutics and Biopharmaceutics, 2012, 81(1): 159-169. DOI:10.1016/j.ejpb.2012.02.004 |
| [25] |
Meng-Lund H, Kasten G, Jensen K T, et al. The Use of Molecular Descriptors in the Development of Co-amorphous Formulations[J]. European Journal of Pharmaceutical Sciences, 2018, 119: 31-38. DOI:10.1016/j.ejps.2018.04.014 |
| [26] |
Pajula K, Lehto V-P, Ketolainen J, et al. Computational Approach for Fast Screening of Small Molecular Candidates to Inhibit Crystallization in Amorphous Drugs[J]. Molecular Pharmaceutics, 2012, 9(10): 2844-2855. DOI:10.1021/mp300135h |
| [27] |
Pajula K, Wittoek L, Lehto V-P, et al. Phase Separation in Coamorphous Systems:In Silico Prediction and the Experimental Challenge of Detection[J]. Molecular Pharmaceutics, 2014, 11(7): 2271-2279. DOI:10.1021/mp400712m |
| [28] |
Lobmann K, Laitinen R, Grohganz H, et al. A Theoretical and Spectroscopic Study of Co-amorphous Naproxen and Indomethacin[J]. International Journal of Pharmaceutics, 2013, 453(1): 80-87. DOI:10.1016/j.ijpharm.2012.05.016 |
| [29] |
Lobmann K, Laitinen R, Grohganz H, et al. Coamorphous Drug Systems:Enhanced Physical Stability and Dissolution Rate of Indomethacin and Naproxen[J]. Molecular Pharmaceutics, 2011, 8(5): 1919-1928. DOI:10.1021/mp2002973 |
| [30] |
Qian S A, Heng W L, Wei Y F, et al. Coamorphous Lurasidone Hydrochloride-Saccharin with Charge-Assisted Hydrogen Bonding Interaction Shows Improved Physical Stability and Enhanced Dissolution with pH-independent Solubility Behavior[J]. Crystal Growth & Design, 2015, 15(6): 2920-2928. |
| [31] |
Dengale S J, Hussen S S, Krishna B S M, et al. Fabrication, Solid State Characterization and Bioavailability Assessment of Stable Binary Amorphous Phases of Ritonavir with Quercetin[J]. European Journal of Pharmaceutics and Biopharmaceutics, 2015, 89: 329-338. DOI:10.1016/j.ejpb.2014.12.025 |
| [32] |
Allesø M, Chieng N, Rehder S, et al. Enhanced Dissolution Rate and Synchronized Release of Drugs in Binary Systems Through Formulation:Amorphous Naproxen-cimetidine Mixtures Prepared by Mechanical Activation[J]. Journal of Controlled Release, 2009, 136(1): 45-53. DOI:10.1016/j.jconrel.2009.01.027 |
| [33] |
Jensen K T, Larsen F H, Cornett C, et al. Formation Mechanism of Coamorphous Drug-amino Acid Mixtures[J]. Molecular Pharmaceutics, 2015, 12(7): 2484-2492. DOI:10.1021/acs.molpharmaceut.5b00295 |
| [34] |
Jensen K T, Lobmann K, Rades T, et al. Improving Coamorphous Drug Formulations by the Addition of the Highly Water Soluble Amino Acid, Proline[J]. Pharmaceutics, 2014, 6(3): 416-435. DOI:10.3390/pharmaceutics6030416 |
| [35] |
Ojarinta R, Lerminiaux L, Laitinen R. Spray Drying of Poorly Soluble Drugs from Aqueous Arginine Solution[J]. International Journal of Pharmaceutics, 2017, 532(1): 289-298. DOI:10.1016/j.ijpharm.2017.09.015 |
| [36] |
Lenz E, Jensen K T, Blaabjerg L I, et al. Solid-state Properties and Dissolution Behaviour of Tablets Containing Co-amorphous Indomethacin-arginine[J]. European Journal of Pharmaceutics and Biopharmaceutics, 2015, 96: 44-52. DOI:10.1016/j.ejpb.2015.07.011 |
| [37] |
Craye G, Lobmann K, Grohganz H, et al. Characterization of Amorphous and Co-Amorphous Simvastatin Formulations Prepared by Spray Drying[J]. Molecules, 2015, 20(12): 21532-21548. DOI:10.3390/molecules201219784 |
| [38] |
Jensen K T, Blaabjerg L I, Lenz E, et al. Preparation and Characterization of Spray-dried Co-amorphous Drug-amino Acid Salts[J]. Journal of Pharmacy and Pharmacology, 2016, 68(5): 615-24. DOI:10.1111/jphp.2016.68.issue-5 |
| [39] |
Mishra J, Rades T, Löbmann K, et al. Influence of Solvent Composition on the Performance of Spray-Dried CoAmorphous Formulations[J]. Pharmaceutics, 2018, 10(2): 47. DOI:10.3390/pharmaceutics10020047 |
| [40] |
Arnfast L, Kamruzzaman M, Löbmann K, et al. Melt Extrusion of High-dose Co-amorphous Drug-drug Combinations[J]. Pharmaceutical Research, 2017, 34(12): 2689-2697. DOI:10.1007/s11095-017-2254-8 |
| [41] |
Lenz E, Lobmann K, Rades T, et al. Hot Melt Extrusion and Spray Drying of Co-amorphous Indomethacin-arginine with Polymers[J]. Journal of Pharmaceutical Sciences, 2017, 106(1): 302-312. DOI:10.1016/j.xphs.2016.09.027 |
| [42] |
Wickström H, Palo M, Rijckaert K, et al. Improvement of Dissolution Rate of Indomethacin by Inkjet Printing[J]. European Journal of Pharmaceutical Sciences, 2015, 75: 91-100. DOI:10.1016/j.ejps.2015.03.009 |
| [43] |
Rask M B, Knopp M M, Olesen N E, et al. Comparison of Two DSC-based Methods to Predict Drug-polymer Solubility[J]. International Journal of Pharmaceutics, 2018, 540(1): 98-105. |
| [44] |
Rams-Baron M, Jachowicz R, Boldyreva E, et al. Amorphous Drug Formulation, in Amorphous Drugs:Benefits and Challenges[M]. 1st ed. New York: Springer International Publishing, 2018.
|
| [45] |
Kissi E, Kasten G, Loebmann K, et al. The Role of Glass Transition Temperatures in Co-amorphous Drug-amino Acid Formulations[J]. Molecular Pharmaceutics, 2018, 15(9): 4247-4256. DOI:10.1021/acs.molpharmaceut.8b00650 |
| [46] |
Lobmann K, Laitinen R, Strachan C, et al. Amino Acids as Co-amorphous Stabilizers for Poorly Water-soluble Drugs-Part 2:Molecular Interactions[J]. European Journal of Pharmaceutics and Biopharmaceutics, 2013, 85(3): 882-888. DOI:10.1016/j.ejpb.2013.03.026 |
| [47] |
Fung M H, Devault M, Kuwata K T, et al. Drug-excipient Interactions:Effect on Molecular Mobility and Physical Stability of Ketoconazole-organic Acid Coamorphous Systems[J]. Molecular Pharmaceutics, 2018, 15(3): 1052-1061. DOI:10.1021/acs.molpharmaceut.7b00932 |
| [48] |
Knapik J, Wojnarowska Z, Grzybowska K, et al. Molecular Dynamics and Physical Stability of Coamorphous Ezetimib and Indapamide Mixtures[J]. Molecular Pharmaceutics, 2015, 12(10): 3610-3619. DOI:10.1021/acs.molpharmaceut.5b00334 |
| [49] |
Dengale S J, Ranjan O P, Hussen S S, et al. Preparation and Characterization of Co-amorphous Ritonavir-indomethacin Systems by Solvent Evaporation Technique:Improved Dissolution Behavior and Physical Stability without Evidence of Intermolecular Interactions[J]. European Journal of Pharmaceutical Sciences, 2014, 62: 57-64. |
| [50] |
Qian S, Heng W, Wei Y, et al. Coamorphous Lurasidone Hydrochloride-Saccharin with Charge-assisted Hydrogen Bonding Interaction Shows Improved Physical Stability and Enhanced Dissolution with pH-Independent Solubility Behavior[J]. Crystal Growth & Design, 2015, 15(6): 2920-2928. |
| [51] |
Paluch K J, McCabe T, Müller-Bunz H, et al. Formation and Physicochemical Properties of Crystalline and Amorphous Salts with Different Stoichiometries Formed between Ciprofloxacin and Succinic Acid[J]. Molecular Pharmaceutics, 2013, 10(10): 3640-3654. DOI:10.1021/mp400127r |
| [52] |
Heikkinen A T, Declerck L, Lobmann, et al. Dissolution Properties of Co-amorphous Drug-amino Acid Formulations in Buffer and Biorelevant Media[J]. Pharmazie, 2015, 70(7): 452-457. |
| [53] |
Ojarinta R, Heikkinen A T, Sievanen E, et al. Dissolution Behavior of Co-amorphous Amino Acid-indomethacin Mixtures:the Ability of Amino Acids to Stabilize the Supersaturated State of Indomethacin[J]. European Journal of Pharmaceutics and Biopharmaceutics, 2017, 112: 85-95. DOI:10.1016/j.ejpb.2016.11.023 |
| [54] |
Petry I, Lobmann K, Grohganz H, et al. Solid State Properties and Drug Release Behavior of Co-amorphous Indomethacin-arginine Tablets Coated with Kollicoat Protect[J]. European Journal of Pharmaceutics and Biopharmaceutics, 2017, 119: 150-160. DOI:10.1016/j.ejpb.2017.06.007 |
| [55] |
Trasi N S, Taylor L S. Dissolution Performance of Binary Amorphous Drug Combinations-impact of a Second Drug on the Maximum Achievable Supersaturation[J]. Int J Pharm, 2015, 496(2): 282-290. DOI:10.1016/j.ijpharm.2015.10.026 |
| [56] |
Teja A, Musmade P B, Khade A B, et al. Simultaneous Improvement of Solubility and Permeability by Fabricating Binary Glassy Materials of Talinolol with Naringin:Solid State Characterization, In-vivo In-situ Evaluation[J]. European Journal of Pharmaceutical Sciences, 2015, 78: 234-244. DOI:10.1016/j.ejps.2015.08.002 |
| [57] |
Wang J, Chang R M, Zhao Y N, et al. Coamorphous Loratadine-citric Acid System with Enhanced Physical Stability and Bioavailability[J]. Aaps Pharmscitech, 2017, 18(7): 2541-2550. DOI:10.1208/s12249-017-0734-0 |
| [58] |
Shayanfar A, Ghavimi H, Hamishekar H, et al. Coamorphous Atorvastatin Calcium to Improve Its Physicochemical and Pharmacokinetic Properties[J]. J Pharm Pharm Sci, 2013, 16(4): 577-587. DOI:10.18433/J3XS4S |
| [59] |
Moinuddin S M, Ruan S, Huang Y, et al. Facile Formation of Co-amorphous Atenolol and Hydrochlorothiazide Mixtures via Cryogenic-milling:Enhanced Physical Stability, Dissolution and Pharmacokinetic Profile[J]. International Journal of Pharmaceutics, 2017, 532(1): 393-400. DOI:10.1016/j.ijpharm.2017.09.020 |