 2019, Vol. 33
2019, Vol. 33 
世界范围内的药品监管,包括美国、欧盟、WHO等均建立符合自身监管实际的监管法规体系框架,明确不同层级监管职责及监管要求。这其中,美国药品监管在检查方面的机构建设、法规发展更具有我国借鉴之处。药品监管效果体现为监管理念、目标的实现程度。回顾美国药品监管百年历程,不难发现任何监管体系均经历从无到有,不断从失败教训中吸取经验,从不成熟到成熟的发展过程。本文通过对美国药品监管体系的设置、发展等特点进行研究和分析,希望为改革过程中的我国药品监管体系提供参考。
1 美国药品监管体系设置在美国,主管药品监督管理的行政机构主要分为两级,即美国食品与药品管理局(U.S. Food and Drug Administration,FDA)和州政府卫生局(一般设有药政机构)。其中,FDA是美国药品安全监管主体,隶属于联邦政府卫生与人类服务部(Department of Health and Human Services,HHS),机构总部由5个办公室承担主要工作职能,分别为食品及兽药监管办公室、全球监管运营及政策办公室、医疗产品及烟草监管办公室、运营办公室和政策、规划、立法及分析办公室,还包括7个专员办公室分别为妇女卫生办公室、少数族裔卫生办公室、首席科学家办公室、执行秘书处办公室、局长顾问办公室、实验室科学与安全办公室、对外事务办公室,共计12个办公室组成。另外,还设有7个中心,分别为食品安全及应用营养中心、兽药中心、国家毒理研究中心、生物制品评价及研究中心(CBER)、烟草产品中心、药品评价及研究中心(CDER)、器械及放射卫生中心。FDA的员工数量超过16000名[1-3]。负责全国范围内食品(家禽类制品除外)、药品、生物制品、医疗器械、放射性产品、化妆品、兽药以及烟草制品等的监管。
FDA的药品生产质量监管具体执行部门主要涉及2个机构,分别为审评部门(CDER、CBER等)、监管实务办公室(Office of Regulatory Affairs,ORA)以及隶属于CDER下的综合质量管理小组(Integrated Quality Assessmment,IQA)。ORA与CDER是平行机构。新药审评、仿制药审评部门与合规办公室综合质量小组均属于CDER的下属部门。审评部门、监管事务办公室及与综合质量管理小组等之间关系详见图 1。

|
图 1 美国FDA药品生产质量监管主要部门设置图示 |
审评部门隶属于医疗产品及烟草监管办公室, 主要包括生物制品评价及研究中心(CBER)、烟草产品中心、药品评价及研究中心(CDER)、器械及放射卫生中心。药物评价和研究中心(CDER)主要包括新药办公室(Office of New Drugs)、仿制药办公室(Office of Generic Drugs)和合规办公室(Office of Compliance)等13个办公室[4]。主要监管处方和非处方药, 新药、仿制药上市前评估, 并负责药物的安全性、质量以及有效性工作。生物制品评价和研究中心(CBER)负责生物学治疗的安全性与有效性, 职责是保证血液、血液制品、疫苗、过敏原制品以及生物治疗产品的安全性和有效性。CDER为FDA最大的职能中心, 有超过4000名员工, 主要工作是对上市前的新药进行审评, 包括新药和仿制药。2008年至2017年, 平均每年有31个新药获批上市[5], 仿制药申请每年约有1000个获批。为缓解审评积压, 近年来CDER人员数量有显著增加, 其中仿制药审评人员数量变化较明显, 如2016年新增125名人员, 仿制药评价办公室达到450人, 最终计划增至630人[6]。2015-2017年CDER人员数量变化详见图 2。

|
图 2 2015-2017年CDER人员数量变化 |
监管实务办公室(Office of Regulatory Affairs,ORA)隶属于全球监管运营及政策办公室,主要由位于马里兰州罗克维尔和马里兰州银泉市的总部办公室以及位于美国各地的地方办事处组成。根据地域和工作需要分设5个区域办公室、20个地方办公室,涉及227个办事处(含设置在国外的办事处)及13个科学实验室[7]。区域办公室是FDA执行事前、事中、事后检查的主要执行部门。每个区域办公室都由区域食品和药品总监(RFDD)领导,区域科学实验室和区域工作人员向RFDD报告。ORA检查范围广泛,主要包括人用和兽用药品、疫苗、医疗器械、化妆品、烟草、食品、防辐射产品等。在美国,消费者每消费1美元,其中有20美分涉及ORA监管的产品[3]。ORA主要职责包括检查及调查(包括监管对象违法犯罪调查)、抽样及分析、进口至美国产品的检查、突发事件处理、召回及强制执行、处理投诉举报等。ORA共有人员5100余人,其中药品相关人员为460余人,约一半是药品GMP检查员。2015-2017年,为适应产品全链条监管要求,ORA人员数量得到大幅提升。2015-2017年ORA人员数量变化详见图 3[3]。

|
图 3 2015-2017年ORA人员数量变化 |
FDA确认药品生产企业是否符合cGMP的现场检查通常可分为四大类:批准前检查、批准后检查、监督cGMP检查、有因调查。CDER与ORA间沟通合作流程参见图 4、图 5、图 6、图 7。2014年以前,根据美国《联邦食品、药品和化妆品法案》(FDCA)规定,每隔2年对药品生产企业进行一次药品GMP常规监督检查。2014年7月,美国国会颁布的《FDA安全和创新法案》(FDASIA)加强了FDA对GMP监管要求,给出了明确的授权要求, 强制应用至相关的供应链管理,要求FDA基于风险对国内外涉及的供应链进行检查。

|
图 4 批准前生产设施检查 |

|
图 5 批准后生产设施检查 |

|
图 6 生产设施有因检查 |

|
图 7 生产设施监督检查 |
为缓解监管人员不足的压力,ORA可能将工作外包给对应的联邦和州政府机构或其他第三方[7]。2017年FDA对监管事务办公室(ORA)进行了大规模重组,地方办公室不再管理各自地理区域的所有产品,而是管理专门的产品领域,并且对于其他地区的该产品领域也承担工作。重组后的每个产品领域都有自己专门针对具体产品法规、科学和市场因素的现场检查机构。通过重组现场检查管理的结构和资源配置,实现从基于地理区域的检查模式转变为聚焦产品质量的检查模式,以利于检查更好地与审评对接。
隶属于CDER的综合质量评估小组(Integrated Quality Assessmment,IQA)、监督办公室(Office of Surveillance,OS)、合规办公室(Office of Compliance,OC)与ORA检查工作相关性较大。为适应药品生产监管的复杂化和全球化要求,提高监管效率,2017年6月,CDER与ORA签署并实施了一项新的操作协议,以实现人用药品审评与现场评估和检查更加全面的整合。该协议的主要举措是组建有审评和现场检查人员共同组成的IQA,以更加全面、细致地考虑所有导致风险的因素。协议适用于批准前、批准后、监督检查以及有因检查,明确了部门职责及工作程序。审评与检查的紧密协作将会增强信息共享、促进双方专业互补、提高监管效率[8]。如注册批准前检查:按照程序要求,由IQA小组评估是否需要实施批准前现场检查,如需检查,由IQA根据对申请生产设施的评估和审查情况(OPF提出)制定检查策略,ORA组织实施,隶属于CDER的药品质量办公室(Office of Pharmaceutical Quality,OPQ)人员参与现场检查, 检查组完成检查报告,IQA小组审查报告并给出建议。常规监督检查由隶属于CDER下的监督办公室(Office of Surveillance,OS)通过风险评估模型(A Risk-based Site Selection Model)选择常规监督检查的场地,ORA安排监督检查[9]。
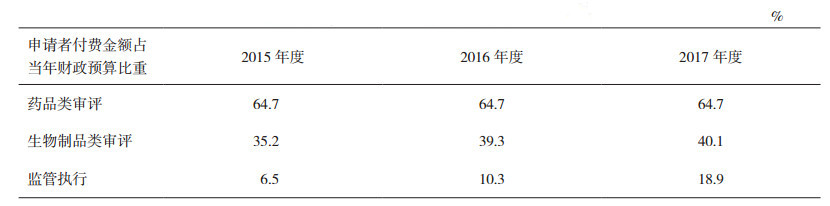
美国国会于1992年审核通过的《处方药申请者费用法案》(Prescription Drug User Fee Act,PDUFA)及2012年通过的《仿制药申请者付费法案》(Generic Drug User Fee Act,GDUFA)要求申请者提交药品上市申请时需缴纳一定的审查费用以抵消FDA药品审评监管的部分成本,并通过增加审评资源,完善审评审批机制,如快速通道、突破性疗法、优先审评、加速审批等方式提高了新药审评的效率。2012年GDUFA的实施促进了消除申请积压,审评队伍得到加强,申报资料质量问题得到改善。根据法案的有关条款,费用主要包括三个部分:申请费(Application Fee)、生产设施费(Establishment Fee)主要用于生产设施检查的费用、产品费(Product Fee)[10]。对费用使用情况和标准依据工作量及复杂程度进行动态调整。上述两法案的实施为FDA药品审评审批及批准后监管工作提供了有力的资源保障,汇总2015-2017年度财政预算及申请者付费所占比重,充分说明了法案在药品监管中的作用,2015-2017年药品审评、监管申请者付费金额比重详见表 1。
|
|
表 1 2015-2017年药品审评、监管申请者付费比重 |
为保证各级监管人员能够接受规范、标准的培训,并使培训效益达到最大化,FDA创建了法规事务办公室大学(ORA U),主要为FDA总部及各州的监管人员提供培训课程,并增加了药品注册、医疗器械、指导文件研讨会等内容。授课方式主要为面授培训、网络培训、广播和光碟培训等。FDA提供超过100个网络课程,包括食品与药品法、公共卫生原则、沟通技巧、食品微生物学、HACCP、食源性疾病调查等[11],并根据不同类型的监管人员提供定制化的网络培训,监管人员通过单独的账户登录,按时按需完成所要求的课程内容,并通过考核获得继续教育学分。
4 思考与展望美国的药品监管通过每一次对《联邦食品、药品和化妆品法案》的修订,对FDA自身、行业及公众产生重大的影响。配套法案制定的大量法规及指南也为规范行业健康发展、保护公众健康做出了巨大贡献,根据历年来的新闻媒体调查显示,FDA长期排在美国公众最受信任的联邦政府机构之列。在宪法的框架下遵循严格的程序,有主要的监管法律,同时有由规制机构制定的相应配套法规,形成完整的监管法律法规体系;依法建立以保证公众健康为目的的独立的监管体系,法规明确了不同监管部门、层级的职责,保证了监管的合法性;以科学为基础,根据监管对象、产品风险合理配置监管资源,强化纵向执法,尽可能避免交叉执法、过度执法,保证法规的有效实施。我国的药品监管改革还在路上,只有先从法律法规层面做好顶层设计,并根据监管形势的需求不断改进,监管机构才能施展开手脚,只有保证人才队伍的质量,才能将政策落实,希望不断壮大我国监管力量,共同推动医药事业的发展[12]。