 2019, Vol. 33
2019, Vol. 33 

2. 国家药品监督管理局药品审评中心, 北京 100022
2. Center for Drug Evaluation, National Medical Products Administration, Beijing 100022, China
2017年6月, 国家药品监督管理局(原国家食品药品监督管理总局)正式加入国际人用药品注册技术要求协调会(ICH), 成为全球第8个监管机构成员, 标志着我国药品标准在国际合作领域迈出重要的一步。而根据ICH发布的协会章程规定, 监管机构成员要逐步实施ICH技术指导原则, 因此, 药物研发的所有参与者(申请人、合同研究机构、监管机构等)均将以ICH指导原则作为行动指南, 实施初期将面临较大的挑战。
尽管科学界对动物致癌性试验, 尤其是啮齿类动物2年致癌性试验的存在价值有很多的争议和讨论, 但是在监管层面达成一致意见之前, 国内的新药申报企业和研究机构仍需按照ICH S1的国际规范全面实施。ICH S1指导原则自从1995年颁布后, 在不断完善, 目前包含三份文件[1-3], 分别是1995年颁布实施的"Guideline on the Need for Carcinogenicity Studies of Pharmaceuticals"(ICH S1A:药物致癌性试验必要性的指导原则)、1997年的"Testing for Carcinogenicity of Pharmaceuticals" (S1B:药物的致癌性试验)和"Dose Selection for Carcinogenicity Studies of Pharmaceuticals"[S1C (R2):药物致癌性试验的剂量选择]。贯彻实施ICH指导原则的前提在于深入了解制定规范的科学基础, 知其然并知其所以然; 结合实际案例的指导原则学习, 将会起到事半功倍的效果。美国食品药品监督管理局(Food and Drug Administration, FDA), 药品评价与研究中心(Center for Drug Evaluation and Research, CDER)近5年内(2014- 2018年)共批准了153个新药申请(New Drug Application, NDA)和60个生物制品许可申请(Biologic Licensing Application, BLA)[4], 本文将对这213个新药进行梳理分析, 从药物致癌性试验的必要性、生物制品致癌性试验的决策、试验类型的选择以及剂量选择等几方面提出一些意见和建议, 以期帮助药物研发参与者加深对ICH S1的理解, 为新药研发和申报工作提供参考。
2 致癌性试验的必要性 2.1 ICH S1A的要求致癌性试验的目的在于发现和识别药物对动物的潜在致瘤性(包括良性、恶性), 继而评价其对人体的相关风险。评估致癌性试验必要性的最基本考虑是临床用药周期长度以及其他安全性研究所发现的相关结果; 其他因素也应被考虑, 如患者人群数量、性别、潜在致癌性的预评估、系统暴露程度、与内源性物质的异同、与临床研究进程相对应的试验时间安排等。
总体来说, 需要进行致癌性试验的情形包括:病人长期应用的药物(关于用药周期在下文讨论), 或频繁的间歇性用药以致总的暴露时间与连续用药类似的药物; 可能导致延长暴露时间的给药药物系统; 同类产品先前已证明有与人相关的致癌性; 构效关系提示有致癌风险的; 在亚慢性或慢性重复给药毒性试验中有不典型增生或癌前病变的; 在组织内长期滞留的母体化合物或其代谢产物导致局部组织增生或其它病理生理变化的; 一些抗肿瘤药物, 拟用于非带瘤病人的辅助治疗或非肿瘤适应症长期使用时也需考虑进行致癌性试验。
不需要进行致癌性试验的情形主要包括:非经常使用或短期暴露的药物(如麻醉药和放射性同位素标记造影剂); 明确有遗传毒性的化合物而临床中仍需长期使用的, 不一定需要进行长期致癌性试验, 但有必要进行长期毒性试验(长达1年)以观察其有无早期致癌作用; 拟定治疗人群的预期寿命较短(如2~3年之内)的药物; 系统暴露量小的局部用药; 对于改盐、改酸根或碱基的药物, 经药代动力学、药效动力学和毒性等方面评估药物暴露量和毒性无明显变化时可不需进行致癌性试验。
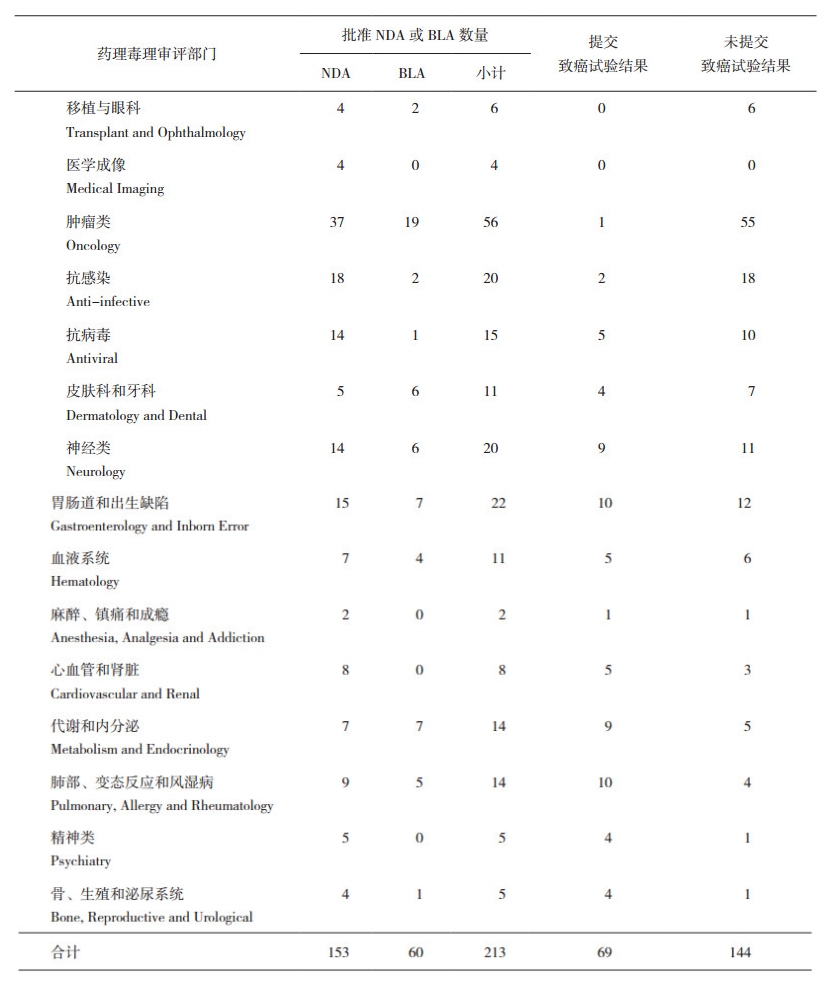
2.2 美国FDA批准的213个新药从表 1和图 1可见, 在上述已批准的213个新药中, 申请上市前完成动物致癌性试验比例较高的几类药物分别是精神类药物, 肺部、变态反应和风湿病药物, 以及代谢和内分泌类药物, 这也是与大家已有的观念相同(即临床长期用药)。以下将从临床用药周期、未进行致癌性试验的原因分类、致癌性试验结果的提交时间、生物制品致癌性试验的决策和不要求而实际进行致癌性试验的情形等5方面进行讨论分析。
|
|
表 1 美国FDA在2014年-2018年期间批准的213项新药分类 |

|
图 1 2014-2018年美国FDA批准新药的致癌性试验开展情况 |
FDA要求, 一般药物使用3个月或更长时间, 需要进行动物致癌性试验。所以在美国大部分长期使用的药物在广泛应用于人体之前, 都已进行了动物致癌性研究。在欧洲, "欧共体药品管理条例"规定需要进行致癌性试验的情况, 包括长期应用的药物, 即至少6个月的连续用药或频繁的间歇性用药以致总的暴露量与前者相似的药物。日本1990年"药物毒性研究指导原则手册"规定:如果临床预期连续用药6个月或更长时间, 则需要进行致癌性试验。尽管连续用药少于6个月, 如果存在潜在致癌性因素, 也可能需要进行致癌性试验。由此可见, 临床用药周期长短是判定致癌性试验必要性的首要考虑因素, 连续用药6个月是一个共识。2015年FDA批准的艾沙康唑(Isavuconazonium, NDA 207500), 用于18岁以上的侵入性曲霉病和毛霉菌病; 申请人引用ICH S1A中的连续用药3个月并依据其他唑类药物已有动物致癌性的报道, 申请豁免动物致癌性试验; 而FDA审评员的观点是大多数连续使用3个月的药物, 用药周期多会达到6个月, 并且要求在药品说明书(Label)中增加"该药未进行2年致癌性试验, 其他唑类药物的小鼠和大鼠致癌性试验中可见肝细胞腺瘤和肝癌", 并须在上市后完成啮齿类动物2年致癌性研究[5]。因此, 在美国FDA的上市后要求(Postmarket Requirements)网页中显示2年小鼠致癌性试验和2年大鼠致癌性试验已在2019年5月完成。
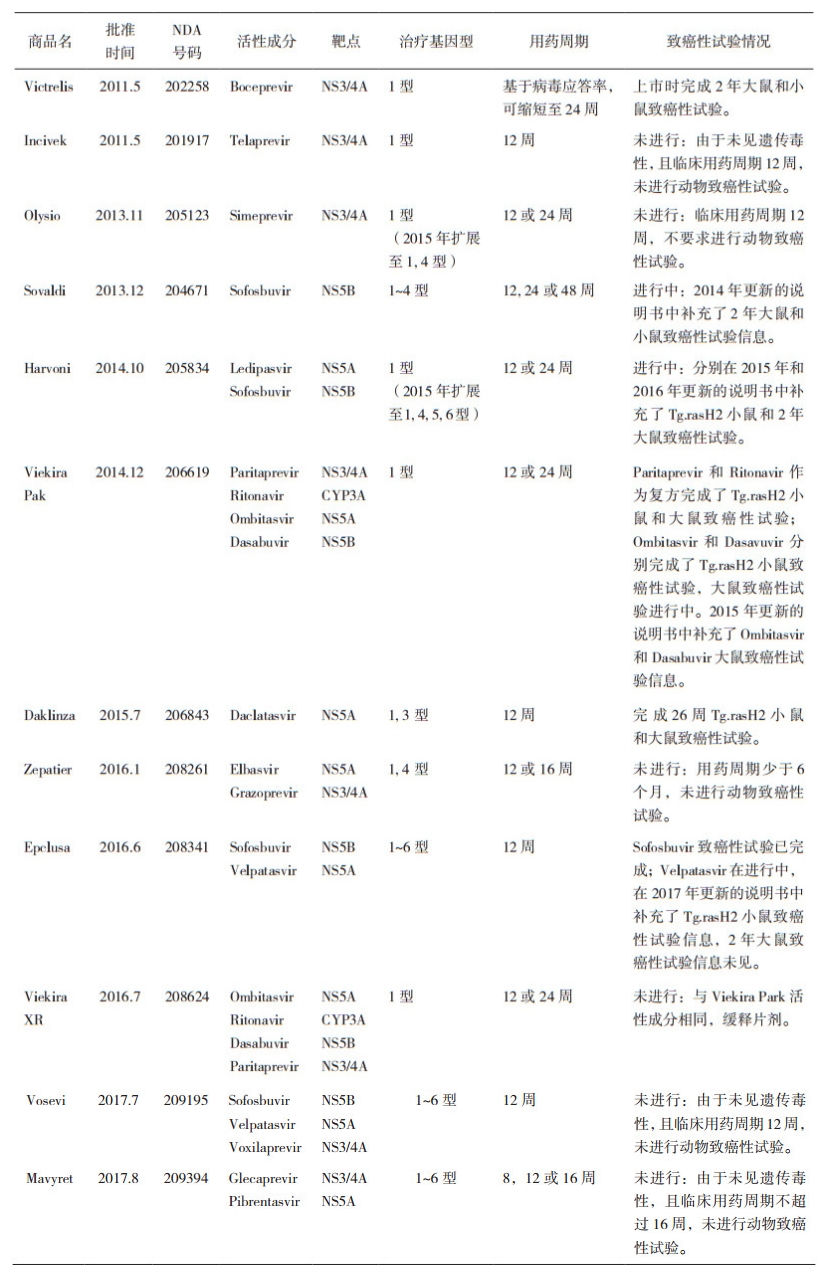
对于用药周期的判定也不是一成不变的, 随着科学界对疾病和药物作用机理认识程度的深入, 用药周期也会有所改变; 用于治疗慢性丙型肝炎(CHC)的直接抗病毒药(DAA)就是其中的典型代表, 随着对丙肝病毒(HCV)复制、生命周期、病毒基因型及病毒蛋白晶型结构认识的深入, 对于该类药物在动物致癌性试验的认识上也在发生着一些改变。自2011年美国FDA批准第一个非结构蛋白(NS3/4A)酶抑制剂至2018年底, FDA共批准了5个单药和7个复方药用于治疗CHC, 其中含有16个新分子实体(New Molecular Entity, NME)。从表 2可见, Viekira XR因与Viekira Pak具有相同的活性成分而除外, FDA批准的11个DAA药物中, 6/11个由于未见遗传毒性, 且临床用药周期少于6个月, 而未进行动物致癌性试验; 其余5/11个都开展了致癌性试验, 但仅2/5个在批准上市时提交了完整的致癌性试验数据, 3/5个在批准上市时未提交或提交部分致癌性试验结果, 但在上市后完成了补充试验。新一代NS3/4A丝氨酸蛋白酶抑制剂单用、复方, 或与干扰素和利巴韦林联用, 临床疗程可缩短至12周, 甚至8周, 且单一疗程就能达到很高的治愈率, FDA接受其不进行动物致癌性试验而批准药物上市[6]。
|
|
表 2 美国FDA批准的不同靶点DAA药物致癌性试验信息归纳 |
将递交NDA时未进行或尚未完成动物致癌性试验的项目简称为无致癌性试验, 将完成至少1项动物致癌性试验的项目简称为有致癌性试验; 从表 3可见, 在过去5年期间, FDA批准的153个NDA项目中, 无致癌性试验的是89个, 有致癌性试验的是64个。无致癌性试验的最常见理由是药物为肿瘤适应症(约42%)或临床用药周期较短(约36%); 还有14/89个是因为系统暴露较低或其他原因(表 3中简称为特殊考虑的); 6/89个药物是获得推迟提交致癌性试验结果的许可(上市后提交), 将在下文另行讨论。
|
|
表 3 美国FDA批准的153个NDA关于动物致癌性试验的分类 |
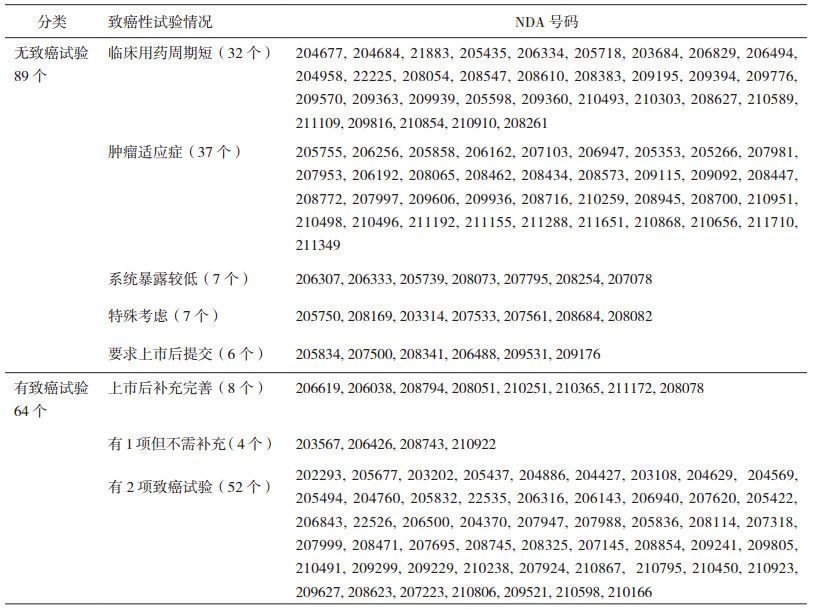
根据ICH S1的要求, 系统暴露量小的局部用药不需要以经口给药途径来评价其对内脏器官的潜在致癌作用。从表 3可见, 非那沙星耳悬液(Finafloxacin Otic Suspension, NDA206307)[7]、Deoxycholic Acid(NDA206333)[8]和Lifitegrast Ophthalmic Solution(NDA208073)[9], 3个药物分别用于治疗急性外耳道炎、成人下颌中重度脂肪堆积和干眼症; Vyzulta(Latanoprostene Bunod Ophthalmic Solution, NDA207795)[10]和Rhopressa (Netarsudil Ophthalmic Solution, NDA208254)[11]均用于治疗开角型青光眼; 上述5个药物均属于局部用药。而Patiromer(NDA205739)[12]和Sodium Zirconium Cyclosilicate(NDA207078)[13], 尽管均属于口服给药用于高钾血症的治疗, 但属于钾离子结合树脂或钾黏结剂, 系统暴露较低, 故未进行动物致癌性研究。
按照临床用药周期, 需要进行动物致癌性试验但没有进行, 且不属于肿瘤适应症或系统暴露较低的药物有7个:Cholbam(Cholic acid, NDA205750)[14], 是胆汁酸的替代治疗, 适应症是罕见胆汁酸合成障碍, 未进行非临床安全性评价动物试验。尿苷三乙酸酯(Uridine Triacetate, NDA208169)[15], 属于嘧啶模拟物, 用于治疗遗传性乳清酸尿症, 尿苷三乙酸酯在体内快速转化为尿苷, 而外源性尿苷已被用于各种适应症, 包括糖尿病肾病、线粒体和神经代谢紊乱, 以及5-氟尿嘧啶毒性, 安全性风险较低。德谷胰岛素(Insulin Degludec, NDA203314)[16], 属于胰岛素类似物, 进行了大鼠重复给药52周的毒性试验, 动物未见药物相关的增生性改变或良性/恶性肿瘤发生。月桂酰阿立哌唑(Aripiprazole Lauroxil, NDA207533)[17], 用于精神分裂症的治疗, 在本品的任何非临床研究中均未见癌前病变, 且已进行口服阿立哌唑的2年小鼠和大鼠致癌性试验, 故未再进行月桂酰阿立哌唑的动物致癌性试验, 但需要在说明书中体现阿立哌唑的动物致癌性试验信息。Genvoya (NDA207561)[18], 为治疗HIV的固定剂量复方制剂, 其中新分子体替诺福韦艾拉酚胺(Tenofovir Alafenamide, TAF)是替诺福韦(Tenofovir, TFV)的前药, 与TFV的另一个前药富马酸替诺福韦酯(Tenofovir Disoproxil Fumarate, TDF)相比, TAF在血浆中更稳定, TAF进入外周单核细胞, 在细胞内代谢为TFV, 并磷酸化为活性代谢产物替诺福韦二磷酸(TFV-DP), 给予TAF可导致HIV靶细胞中TFV-DP水平升高, 同时降低TFV的循环水平, 有望降低TFV的脱靶效应, 提高安全性。由于TAF在小鼠和大鼠体内可快速转化为TFV, 因此, 未进行TAF的致癌性研究。Emflaza(Deflazacort, NDA208684)[19], 属于皮质类固醇药物前体, 用于治疗杜氏肌营养不良, 由于啮齿动物不能预测免疫抑制剂类药物(包括糖皮质激素)与人体相关的肿瘤类型, FDA同意豁免2年大鼠致癌性试验。Deutetrabenazine(NDA208082)[20], 美国FDA批准的首个氘代药物, 是四苯喹嗪(Tetrabenazine)的氘代产物, 药代动力学特征在氘代以后得到改善, 半衰期明显延长, 从而可以使用更低的治疗剂量; 由于在Tetrabenazine给予p53+/-小鼠26周的致癌性试验中未见肿瘤发生率增加, 故针对本品未进行致癌性研究。
2.2.3 致癌性试验结果的提交时间当需要进行致癌性试验时, FDA要求申请人在递交NDA之前或递交NDA同时提供致癌性试验研究结果。若对患者人群存在特殊担忧, 在进行大样本临床试验之前需完成啮齿类动物的致癌性试验[21]。对于开发用于治疗某些尚无有效治疗手段的严重疾病的药物, 或用于老龄病人群体的药物, 申请上市前可不必完成动物致癌性试验, 但要求在上市后伴随4期临床进行致癌性试验。
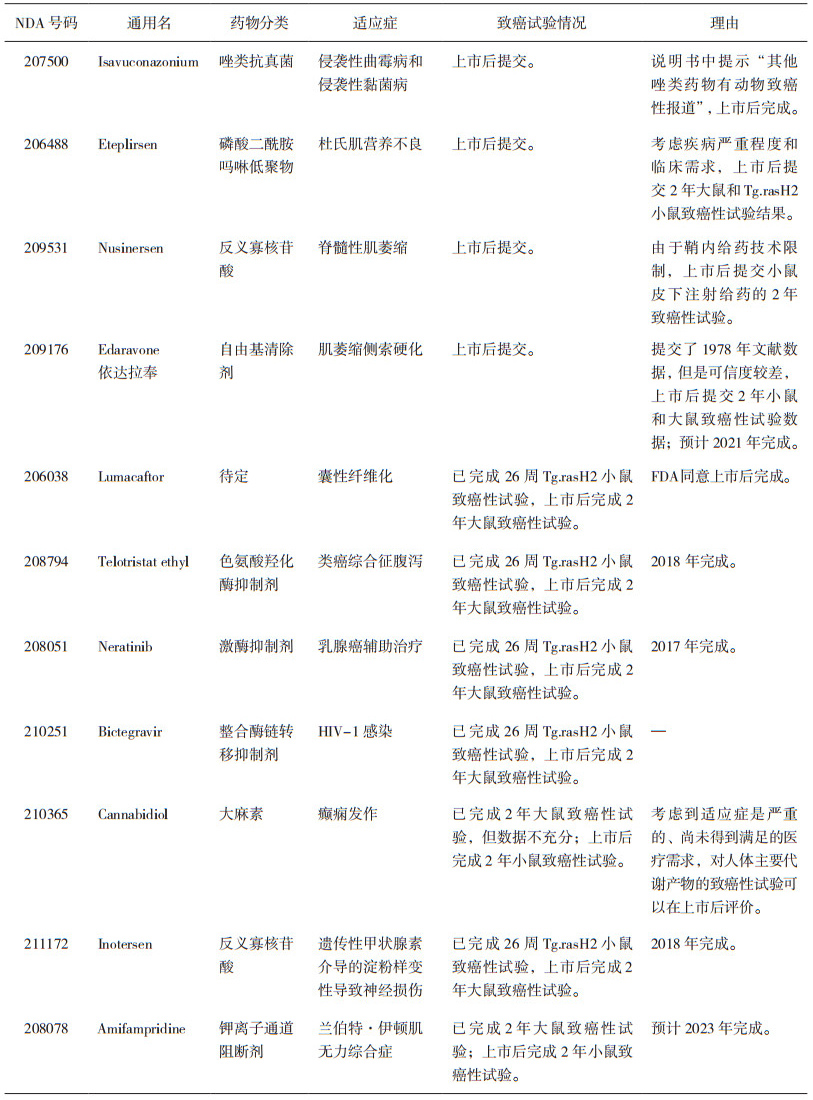
从药物开发成本的角度考虑, 为获得最大收益, 申请人也希望在上市后提交或补充完善动物致癌性试验资料; 过去5年期间, 共有14个药物存在这样的情况, 其中的3个治疗丙肝的药物在前文已讨论, 从表 4所列的11个药物可见:6/11个是神经类药物, 1/11个是针对囊性纤维化的药物, 还有1/11个是针对HIV感染的药物。FDA会根据每个药物的自身特点, 权衡临床获益与风险管控, 特别是对适应症较为严重、医疗需求尚未得到满足的新药, 会批准其上市后进行或补充完善致癌性试验资料。
|
|
表 4 上市后提交或上市后补充完善的案例 |
ICH S1中针对生物制品(大分子药物)的主要要求:对于一些经化学合成、由动物或人体来源提取纯化或通过生物技术方法生产的内源性肽类或蛋白质类物质及其类似物, 所产生的生物作用与天然产物明显不同, 或经过修饰导致产品结构与天然产物相比有明显变化, 或在人体局部或全身的浓度明显增加(即达到药理学水平)时应考虑进行致癌性试验; 而作为替代性治疗的内源性物质, 且浓度在生理水平时可不需进行致癌性试验[1]。
对于生物制品(大分子药物)是否进行致癌性试验的早期决策路线是过于简单的, 主要两点考虑, 其一是临床用药周期, 其二是啮齿动物致癌性研究的可行性, 包括是否在动物体内产生药理活性, 以及是否产生中和抗体。但针对生物制品的决策路线, 如今新的倾向[22]是除了临床用药周期外, 还要考虑风险担忧, 包括是否属于生长因子类、是否存在免疫抑制作用等, 如果没有这些方面的担忧, 只需书面说明不进行动物致癌性试验的理由; 如果存在以上担忧, 就需进一步采取证据权重(Weight of Evidence)分析, 分析内容主要包括以下方面:(1)靶点作用是否与肿瘤相关; (2)是否可采用基因敲除小鼠来说明致癌性特征; (3)是否可从肿瘤促进模型中获得信息; (4)是否可获得体外细胞增殖试验; (5)人体基因变异与靶点调控作用的相似性; (6)进行2年啮齿动物致癌性试验是否可降低风险; (7)患者的风险和获益考虑; (8)是否可通过说明书或上市后监管来控制风险。申请人应根据以上几点提出书面依据。IL-17A单抗(Ixekizumab, BLA125521)[23]关于致癌性试验的权重分析方法就是一个很好的参考, 简要来说:从物质基础上来看, 该单抗是大分子蛋白, 不进入细胞核, 不与DNA相互作用。前期毒理试验结果, 在非人灵长类毒性试验中, 对淋巴细胞亚群、NK细胞功能均无改变, 不影响动物的T细胞依赖性抗体反应(TDAR), 淋巴器官无病变。从靶点调节与肿瘤的相关性上来看, 没有确切结论, IL-17A有明确促炎作用, 而慢性炎症与肿瘤的相关性也早已得到证实, 本品是IL-17A拮抗剂, 有一定的抑制炎症作用; 但是也有研究显示IL-17A有抗肿瘤作用; 即有研究表明, 采用IL17缺陷小鼠可见肿瘤生长加快或转移现象。体外试验研究, IL-17A可促进细胞增殖。临床报道显示, IL17缺陷个体易发口腔念珠菌感染。本品在正常啮齿动物体内不具有药理学活性。从风险获益角度考虑, 本品用于中重度斑块性银屑病和活动性银屑病关节炎。总体来说, IL17的抑制可以为肿瘤生长创造不利环境, 可通过上市后监测提供最终风险评估。审评人员最终同意豁免啮齿动物致癌性试验研究。
过去5年, FDA批准的60个大分子药物中, 绝大多数(55/60个)未进行啮齿动物致癌性试验, 仅有5/60个药物进行了1项或2项动物致癌性试验(详见表 5)。2个GLP-1受体激动剂(Dulaglutide和Semaglutide)[24-25], 与其他已上市的同靶点药物相似, 具有甲状腺肿瘤的担忧, 且2个药物在啮齿动物体内具有明确的药理学作用, 而免疫原性较弱。Natpara是重组人甲状旁腺激素, 在大鼠致癌性试验中可见骨肉瘤发生率的增加, 且具有给药剂量和给药时间的相关性, 骨肿瘤的人体风险不能排除; 因此, 在该药品的说明书中, 不仅有关于骨肿瘤的黑框警告, 并且该药的使用还纳入了风险评估和减低策略(Risk Evaluation and Mitigation Strategies, REMS), 以此来确保药品获益大于风险[26]。瑞利珠单抗(Reslizumab)是一个针对IL-5的人源化单抗(IgG4 kappa), 即IL-5拮抗剂; 临床前研究显示, 该产品的药理作用相关动物包括小鼠、兔和猴, 而亲和力数据显示, 人、猴、小鼠的KD值分别是24、20和31pM, 在小鼠重复给药6个月的毒性试验结果显示, 随着给药时间的延长, 系统暴露量降低, 表明小鼠体内有中和抗体产生。因此, 该产品最终是采用Tg.rasH2小鼠进行26周致癌性试验替代野生型小鼠的2年致癌性试验, 且豁免2年大鼠致癌性试验[27]。
|
|
表 5 BLA申请中包含啮齿动物致癌性试验的情形 |
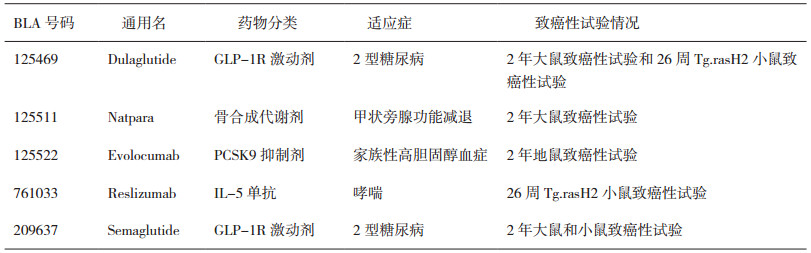
有趣的是, FDA在同一年批准了2个PCSK9 (Proprotein Convertase Subtilisin Kexin Type 9)抑制剂[28-29], 除了表 5所列的Amgen公司的Evolocumab, 另一个是S a n o f i-A v e n t i s公司的A l i r o c u m a b (BLA125559);然而, Evolocumab进行了2年地鼠致癌性试验, 而Alirocumab未进行动物致癌性试验。针对Alirocumab, 尽管采用大鼠进行致癌性试验是可行的, 但申请人依据前期毒理研究结果和文献报道申请豁免致癌性试验:基于文献, 可获得PCSK9抑制剂与肿瘤不具有相关性, 降低胆固醇可通过改变肠道胆汁酸负荷或抑制免疫系统功能来影响肿瘤风险的证据缺乏; 基于研究数据, PCSK9抑制后与免疫抑制无关联性, PCSK9抑制与肠道胆汁酸负荷之间无关联性, 在毒理研究项目中均未见肿瘤或癌前病变。血浆胆固醇与大多数肿瘤风险之间存在惊人的负相关, 最有可能的解释是所谓的临床前肿瘤效应。大量的证据支持这样一种假设, 即低胆固醇水平虽然与总体肿瘤风险增加有关, 但不太可能是这种风险增加的原因。针对Evolocumab, 为解决相似的风险担忧, 进行了2年地鼠致癌性试验, 每2周给药1次, 剂量达到100 mg·kg-1, 皮下注射, 试验期间可见总胆固醇和低密度脂蛋白胆固醇降低, 未见药物相关肿瘤, 与临床人体剂量相比(140 mg Q2W, 420 mg QM和420 mg Q2W), 暴露分别增加了38、15和6.6倍。
2.2.5 不要求但实际进行了致癌性试验的情形从进行致癌性试验的69个药物中, 可以看到一些药物尽管进行了动物致癌性试验, 但是从临床应用周期来看, 认为是不需要进行动物致癌性试验的, 且这些致癌性试验的结果在安全性评估中也没有显示出价值。具体包括:帕拉米韦(Peramivir, NDA206426)[30], 是一种流感病毒神经氨酸酶抑制剂, 静脉给药用于治疗2岁及以上出现症状不超过2天的急性非复杂流感, 进行了2年大鼠致癌性试验, 而2年小鼠致癌性试验因原研究单位(Johnson & Johnson)停止开发而试验中止(未进行病理学检查); 说明书中显示"未进行静脉注射给药的致癌性研究, 进行了大鼠口服给药2年的致癌性研究, 且未见药物相关肿瘤发生"。Defibrotide (NDA208114)[31], 去纤苷酸, 是从猪的肠道组织中提取的一种以单链聚脱氧核糖核酸复合物为主的钠盐, 静脉输注给药用于治疗造血干细胞移植后发生的重度肝小静脉闭塞症, 最长给药周期是60天; 但是, 在较早的年代(1989-1990年)进行了拌食给予大鼠和小鼠的2年致癌性试验, 而在说明书中描述为"未进行静脉给药的致癌性研究"。Avatrombopag(NDA210238)[32], 促血小板生成素受体激动剂, 临床口服给药用于血小板减少症的治疗, 最长给药周期是13天, 但是仍进行了2年大鼠和2年小鼠致癌性试验, 尽管动物可见类胃癌发生率的增加, 但是认为类胃癌可能是由于长时间的高胃泌素血症所致, 且高胃泌素相关的类胃癌在啮齿动物中通常被认为是低风险或与人类相关性较低。Moxidectin(NDA210867)[33], 一种驱虫剂(盘尾丝虫), 临床上口服单次给药, 非临床试验中仍进行了拌食给药的2年大鼠和小鼠致癌性试验, 因FDA统计部门未完成致癌性试验数据的统计分析, 说明书中仍是"未确认长期致癌效应"。Tafenoquine(NDA210795)[34], 是一种抗疟药, 用于间日疟原虫的根治(预防复发); 临床单次用药; 进行了2年小鼠和大鼠致癌性试验, 说明书中显示"由于临床中单次用药, 动物试验的结果不能代表人体致癌性风险"。Lofexidine (NDA209229)[35], 中枢α2肾上腺素能激动剂, 用于减轻阿片类药物戒断症状, 临床最长给药周期是14天, 同样也是在较早的年代(1977-1978年)开展了2年大鼠和小鼠致癌性试验, 但是均因未执行GLP、未测定饲料中药物含量以及高剂量不充分或动物死亡较多等原因认定2项试验无效; 说明书中显示"目前还没有足够的长期动物试验来评估该产品的潜在致癌性"。
3 致癌性试验类型的选择 3.1 ICH S1B的要求药物致癌性试验仅在获得一定的关键信息后才进行, 包括遗传毒性研究的结果、用药人群、临床用药方案、动物和人体药效动力学(选择性、剂量-反应关系)以及重复给药毒性试验结果。如果任何种属(包括非啮齿动物)的重复给药毒性试验可能表明受试物具有免疫抑制作用、激素活性或其他被认为对人体是一种危险因素的活性, 那么这类信息就应在进一步评价潜在致癌性的试验设计中予以考虑。
选择具体致癌性试验方法时应具体问题具体分析。鉴于致癌过程的复杂性, 任何单一的试验方法都无法预测所有人用药物的潜在致癌性。基本方案包括一项长期啮齿类动物致癌性试验, 加上另一项附加的体内致癌性试验作为补充, 以提供长期致癌性试验不易得到的其他信息; 除非是短期或长期致癌性试验和遗传毒性试验以及其他数据提示一种药物明确对人类具有致癌风险, 通常不用再次进行致癌性试验。而附加的体内致癌性试验包括:短期或中期啮齿类动物体内试验系统(应尽量使用能提供致癌终点的体内模型, 包括啮齿类启动-促进模型、转基因鼠致癌模型或新生啮齿类动物致癌模型), 以及第二种属啮齿类动物长期致癌性试验。
长期致癌性试验的动物种属选择时, 应考虑以下因素:药理学、重复给药毒性、受试物的代谢特性、毒代动力学和给药途径, 在缺乏确凿证据时, 推荐选择大鼠。
3.2 FDA批准的127项致癌性试验分析过去5年期间, FDA批准的213个NDA或BLA中, 共有69个新药完成了127项动物致癌性试验(详见表 6)。与Wistar大鼠相比, 尽管SD大鼠存在2年生存率偏低的情况, 但在60项2年大鼠致癌性试验中, 46/60项试验仍是采用了SD大鼠; 这可能与该品系动物背景数据较为充足, 且重复给药毒性试验中多采用它有关。而同一个品系的SD大鼠又可以细分为Crl:CD(SD)和Hsd:SD等不同种群, 不同种群的大鼠在自发性肿瘤发生率甚至肿瘤类型上也存在轻微差别[36-37], 需要在动物选择时给予关注。理想条件下应采用与人类代谢特征尽可能相似的啮齿类动物品系。在普通小鼠的2年致癌性试验中, 更多的是采用封闭群CD-1小鼠, 较少采用杂交系小鼠。唯一的新生小鼠致癌性试验, 是采用Swiss小鼠给药1年的方式。在ICH S1B中推荐的转基因模型包括p53+/-缺失模型、TgAC模型、TgHras2模型(或称Tg.rasH2小鼠模型)和XPA缺失模型, 但是这些模型普遍有如下缺陷:(1)不能100%检测出人类致癌剂, 无法明确区分鼠类致癌剂非人类致癌剂与人类致癌剂非鼠类致癌剂; (2)预测致癌剂的人类靶器官方面应用有限, 如肝脏[38-39]; (3)对基因毒性和非基因毒性致癌物的反应不一致(如TgAC和p53+/-缺失模型); (4)定量风险预测方面的使用范围有限[40]; (5)替代模型的遗传背景对肿瘤的发生造成了影响[41], 如C57BL/6背景对肝癌有抑制作用, 导致该遗传背景的模型对某些致癌剂不敏感。而每个模型小鼠均有其优缺点; Tg.rasH2小鼠模型由于针对基因毒性和非基因毒性致癌剂均有较高的检出率, 且对人类致癌剂非鼠类致癌剂较敏感[42-43], 在目前的新药非临床研究中, 较多采用的是"2年大鼠致癌性试验+26周Tg.rasH2小鼠致癌性试验"这种组合方式。需要提醒的是, 由于药物种类不同, 药理学特性及体内代谢特点不同, 临床应用上的诸多差异, 在选择具体的致癌性试验类型时, 例如采用常规大、小鼠的2年生命周期给药, 还是采用上述的"2年大鼠致癌性试验+26周Tg.rasH2小鼠替代性致癌性试验"的组合方式, 必须在致癌性试验"特殊方案评估(Special Protocol Assessment, SPA)"过程中获得FDA的认可或建议。关于FDA致癌性研究SPA的指导文件以及FDA内部致癌性试验评估执行委员会(Executive Carcinogenicity Assessment Committee, E C A C)的功能和运作, 将在后续专题文章中详述。
|
|
表 6 近5年FDA批准新药的致癌性试验类型信息 |

关于高剂量的选择, ICH S1C(R2)推荐的指标包括[3]:(1)基于毒性改变的指标:ICH安全性专家小组同意继续使用最大耐受剂量(MTD)作为有用的致癌性试验选择高剂量的毒性指标, 该剂量是指致癌性试验中预期产生可耐受的最小毒性作用的剂量, 包括与对照组相比体重的增长率减少不超过10%、有靶器官毒性、临床病理参数的明显改变等; (2)药代动力学指标:测定血浆中游离药物浓度是考察组织中游离药物浓度最合适的间接测定方法, 而AUC是最全面的药代动力学指标, 兼顾了化合物的血浆浓度和体内滞留时间, 而基于以MTD进行的致癌性试验数据库的分析, 以啮齿动物血浆中原型药物和/或代谢产物AUC为人的25倍作为致癌性试验的高剂量被认为是实用的; 特别注意的是要有药物在啮齿动物与人体中代谢相似的证据, 以及在估计相对暴露量时, 应考虑不同种属之间蛋白结合率的差异; (3)吸收饱和量:可用药物或其活性代谢物系统生物利用度计算得到的吸收饱和量作为致癌性试验的高剂量, 同时低、中剂量也需考虑药物代谢的饱和以及消除途径; (4)药效学指标:选择的高剂量应使动物产生的药效学反应足够大, 以避免需要进一步递增剂量, 然而, 该剂量不应干扰生理或体内平衡状态, 从而降低研究的价值, 如造成低血压和抑制凝血等; (5)最大可行剂量:如果无法获得MTD或无法达到25倍人体暴露量AUC时, 各个药物监管机构均以最大可行剂量(MFD)作为可接受的指标。通过拌食法进行致癌性试验时, 最大量是饲料量的5%;当用其他途径时, 还应考虑到可行性和局部耐受性; (6)剂量限度:当推荐人用最大剂量不超过500 mg·d-1时, 致癌性试验的高剂量可设定在1500 mg·kg-1·d-1, 但是, 需要该剂量水平的系统暴露量比人体拟定治疗剂量所能达到的暴露量至少高一个数量级(否则应考虑增加暴露量或重新考虑动物模型); 如果人用剂量超过500 mg·d-1, 高剂量还可增加至MFD。
关于中、低剂量, 通常以倍率递减方式来确定, 以期获得剂量相关的致癌性反应(如果药物存在致癌性的话)。
4.2 2年大鼠致癌性试验剂量选择的实例分析从FDA批准的54项2年大鼠致癌性试验数据来看:绝大多数药物(48/54)是依据MTD, 或AUC比值, 或二者的综合分析来确定高剂量组给药剂量; 4/54项试验是依据MFD; 1/54项试验是依据药效学作用; 还有1/54项是采用六大标准之外的方法(局部药物浓度)。剂量选择的标准大多不是唯一性, 利用多种剂量选择标准, 为设计最佳的药物致癌性试验提供了更大的灵活性。因篇幅有限, 本文仅将有代表性的几个剂量选择方式列举如下:
Evolocumab(BLA125522), PCSK9单抗, 临床给药途径是皮下注射, 人体最大推荐剂量(MRHD)是140 mg每两周一次或420 mg每月一次。在地鼠重复皮下注射给药13周的毒性试验中, 给药剂量分别是100 mg·kg-1和300 mg·kg-1; 结果显示, 在100 mg·kg-1水平, 动物血清CHOL、HDL和LDL水平均达到最大抑制效应; 因此, 在2年致癌性试验中, 采用皮下注射给药, Q2W, 剂量分别是10、30和100 mg·kg-1; 结果显示未见药物相关肿瘤发生。
Plecanatide(SP-304, NDA208745)[44], 为含有16个氨基酸的化学合成多肽, 鸟苷酸环化酶C激动剂, 局部作用于肠上皮表面, 促进肠液分泌, 用于治疗慢性特发性便秘, MRHD是3 mg·d-1。FDA的ECAC关于剂量设置的建议:基于300 mg·kg-1·d-1组动物有体重增长量降低, 推荐雌性动物剂量是10、30和100 mg·kg-1·d-1; 基于大鼠肠道局部药物浓度较大, 达到了最大药理学作用, 推荐雄性动物剂量是10、30和100 mg·kg-1·d-1。
巴瑞替尼(Baricitinib, NDA207924)[45], JAK1/2激酶抑制剂, 用于治疗类风湿关节炎, MRHD是2 mg·d-1。在大鼠重复灌胃给药26周的毒性试验中, 雄性剂量分别是0.5、5、25和100 mg· kg-1·d-1, 其中, 5和25 mg·kg-1·d-1组动物体重分别降低8%和16%;雌性剂量分别是0.5、5、25和100/60 mg·kg-1·d-1, 其中, 25 mg·kg-1·d-1组动物体重无降低, 而外周血淋巴细胞降低39%。根据以上信息, 2年大鼠致癌性试验雄性动物剂量选择1、3和8 mg·kg-1·d-1, 雌性动物剂量是3、8和25 mg·kg-1·d-1。
4.3 短期致癌性试验的剂量选择的实例分析与2年大鼠致癌性试验不同, 转基因小鼠致癌性试验更关注的是动物耐受性, 即基于毒性改变来选择高剂量, 而非体内暴露量[46-47]。因此, 在FDA最近5年期间批准的21项Tg.rasH2小鼠致癌性试验中(详见表 7), 有2项试验的给药剂量均大于或等于剂量限度法的1500 mg·kg-1·d-1, 分别是Dasabuvir(NDA206619)和Lumacaftor (NDA206038)。且与大鼠致癌性试验结果的描述方式不同, 在药品说明书中, 一般也不描述转基因小鼠致癌性试验中动物体内暴露量与人体暴露量的比值或倍数。
|
|
表 7 过去5年期间FDA批准的21项Tg.rasH2小鼠致癌性试验剂量设置信息 |
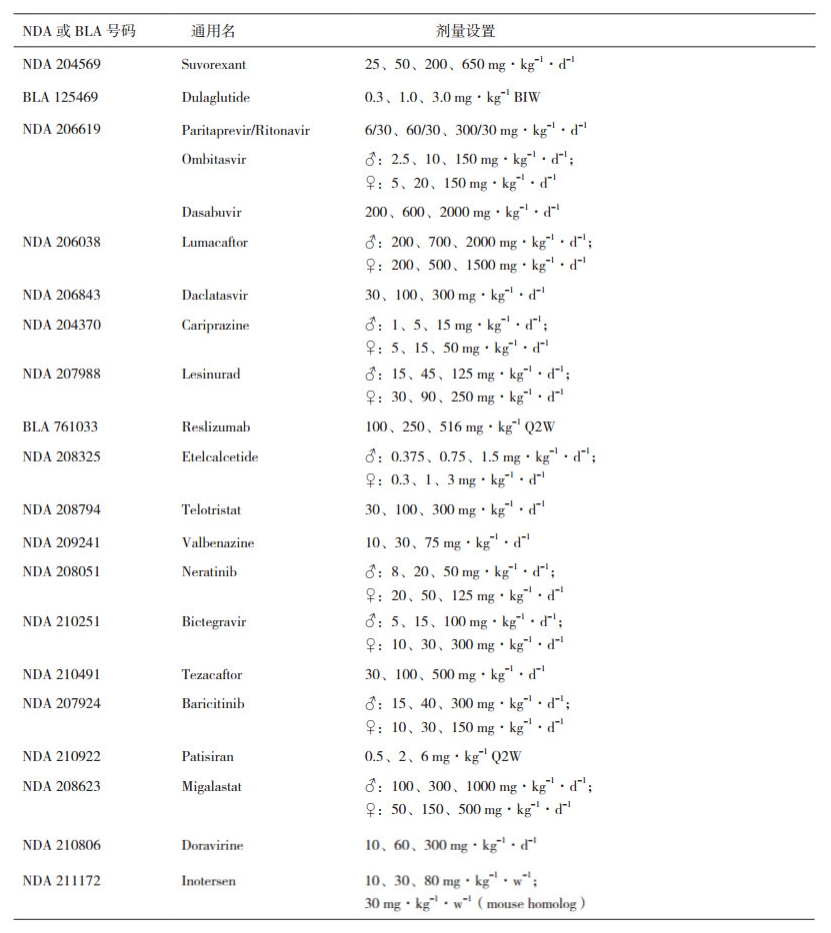
26周Tg.rasH2小鼠致癌性试验的剂量选择均依据Non-Tg.rasH2小鼠4周剂量探索试验的结果, 同样以Baricitinib为例[45], 在探索试验中, 雄性和雌性动物给药剂量均是75、150和300 mg·kg-1·d-1; 根据300 mg·kg-1剂量水平, 雄性动物淋巴细胞降低50%, 雌性动物出现的肾小管变性/坏死、扩张, 判定雄性和雌性动物的MTD分别为300和150 mg·kg-1·d-1。故在26周致癌性试验中, 雄性动物给药剂量分别是15、40和300 mg·kg-1·d-1, 雌性动物给药剂量分别是10、30和150 mg·kg-1·d-1。结果显示各组动物均未见药物相关肿瘤发生。
5 结论啮齿类动物致癌性试验的试验周期长、费用高, 且致癌性试验的复杂程度远远超出指导原则的要求, 药品申请人需根据拟定的临床用药周期、药物代谢特征、药理毒理学研究结果等综合考虑动物致癌性试验的必要性; 结合临床试验进度和非临床研究机构的综合能力合理选择致癌性试验的启动时机。对于大分子蛋白等生物制品, 也不能想当然地认为不需开展动物致癌性试验, 更应该是结合产品特点进行证据权重分析, 理性评估。当前动物致癌性试验的常规组合方式是2年大鼠致癌性试验加上一项26周Tg.rasH2小鼠致癌性试验, 但是, 两项试验在试验设计、饲养条件、剂量选择、统计分析等方面均存在区别, 研究者要结合实际案例加强指导原则学习。在进行致癌性试验决策时, 除了ICH S1指导原则外, 还要兼顾ICH M3和ICH S6等指导原则中关于致癌性试验的相关考虑。致癌性试验不同于任何其他非临床安全性研究, 药物监管机构也会介入方案评估, 尤其是试验类型、剂量设计等, FDA的ECAC会给予申请人最终的方案评估意见和建议, 我国也鼓励申请人就致癌性试验方案与国家药品监督管理局药品审评中心进行沟通交流。因此, 申请人应严格按照法规要求与监管部门加强沟通交流, 取得监管部门对方案评估的建议和指导, 在实施ICH S1指导原则的过程中不断积累和完善经验。另外, 目前ICH S1(R1)专家工作组正在针对致癌性试验策略对S1指导原则进行修订, 建议申请人和研究者密切关注ICH官网(www.ich.org/products/guidelines.html)公布的相关进展。
致谢
感谢军事医学科学院毒物药物研究所的廖明阳教授对本文的指导和帮助!
| [1] |
ICH. S1A Guidance for Industry: The Need for Long-term Rodent Carcinogenicity Studies of Pharmaceuticals[S]. 1996.
|
| [2] |
ICH. S1B Guidance for Industry: Testing for Carcinogenicity of Pharmaceuticals[S]. 1997.
|
| [3] |
ICH. S1C (R2) Guidance for Industry: Dose Selection for Carcinogenicity Studies of Pharmaceuticals[S]. 1997.
|
| [4] |
FDA. Drug Approval Report[EB/OL].[2019-07-27]. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/.
|
| [5] |
FDA. Pharmacology/Toxicology Review: Isavuconazonium[R]. NDA 207500. CDER, 2015.
|
| [6] |
于春荣, 笪红远, 王庆利. 直接抗丙肝病毒新药的致癌性研究评价[J]. 中国新药杂志, 2017, 26(1): 32-35. |
| [7] |
FDA. Pharmacology/Toxicology Review: Finafloxacin Otic Suspension[R]. NDA 206307. CDER, 2014.
|
| [8] |
FDA. Pharmacology/Toxicology Review: Deoxycholic Acid[R]. NDA 206333. CDER, 2015.
|
| [9] |
FDA. Pharmacology/Toxicology Review: Lifitegrast Ophthalmic Solution[R]. NDA 208073. CDER, 2016.
|
| [10] |
FDA. Pharmacology/Toxicology Review: Vyzulta[R]. NDA 207795. CDER, 2017.
|
| [11] |
FDA. Pharmacology/Toxicology Review: Rhopressa[R]. NDA 208254. CDER, 2017.
|
| [12] |
FDA. Pharmacology/Toxicology Review: Patiromer[R]. NDA 205739. CDER, 2015.
|
| [13] |
FDA. Pharmacology/Toxicology Review: Sodium Zirconium Cyclosilicate[R]. NDA 207078. CDER, 2018.
|
| [14] |
FDA. Pharmacology/Toxicology Review: Cholbam[R]. NDA 205750. CDER, 2015.
|
| [15] |
FDA. Pharmacology/Toxicology Review: Uridine Triacetate[R]. NDA 208169. CDER, 2015.
|
| [16] |
FDA. Pharmacology/Toxicology Review: Insulin Degludec[R]. NDA 203314. CDER, 2015.
|
| [17] |
FDA. Pharmacology/Toxicology Review: Aripiprazole Lauroxil[R]. NDA 207533. CDER, 2015.
|
| [18] |
FDA. Pharmacology/Toxicology Review: Genvoya[R]. NDA 207561. CDER, 2015.
|
| [19] |
FDA. Pharmacology/Toxicology Review: Emflaza[R]. NDA 208684. CDER, 2017.
|
| [20] |
FDA. Pharmacology/Toxicology Review: Deutetrabenazine[R]. NDA 208082. CDER, 2017.
|
| [21] |
ICH. M3(R2) Guidance on Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals[S]. 2009.
|
| [22] |
ICH. S6(R1) Preclinical Safety Evaluation of Biotechnologyderived Pharmaceuticals[S]. 2011.
|
| [23] |
FDA. Pharmacology/Toxicology Review: Ixekizumab[R]. BLA125521. CDER, 2016.
|
| [24] |
FDA. Pharmacology/Toxicology Review: Dulaglutide[R]. BLA125469. CDER, 2014.
|
| [25] |
FDA. Pharmacology/Toxicology Review: Semaglutide[R]. BLA125511. CDER, 2015.
|
| [26] |
FDA. Pharmacology/Toxicology Review: Natpara[R]. BLA209637. CDER, 2017.
|
| [27] |
FDA. Pharmacology/Toxicology Review: Reslizumab[R]. BLA761033. CDER, 2016.
|
| [28] |
FDA. Pharmacology/Toxicology Review: Evolocumab[R]. BLA125522. CDER, 2015.
|
| [29] |
FDA. Pharmacology/Toxicology Review: Alirocumab[R]. BLA125559. CDER, 2015.
|
| [30] |
FDA. Pharmacology/Toxicology Review: Peramivir[R]. NDA 206426. CDER, 2014.
|
| [31] |
FDA. Pharmacology/Toxicology Review: Defibrotide[R]. NDA 208114. CDER, 2016.
|
| [32] |
FDA. Pharmacology/Toxicology Review: Avatrombopag[R]. NDA 210238. CDER, 2018.
|
| [33] |
FDA. Pharmacology/Toxicology Review: Moxidectin[R]. NDA 210867. CDER, 2018.
|
| [34] |
FDA. Pharmacology/Toxicology Review: Tafenoquine[R]. NDA 210795. CDER, 2018.
|
| [35] |
FDA. Pharmacology/Toxicology Review: Lofexidine[R]. NDA 209229. CDER, 2018.
|
| [36] |
张素才, 芮志佩, 张冬霞, 等. 长期致癌试验动物饲养管理技术关注要点[J]. 中国新药杂志, 2017, 26(2): 162-168. |
| [37] |
Brower M, Grace M, Kotz C, et al. Comparative Analysis of Growth Characteristics of Sprague Dawley Rats Obtained from Different Sources[J]. Lab Anim Res, 2015, 31(4): 166-173. DOI:10.5625/lar.2015.31.4.166 |
| [38] |
Gulezian D, Jacobson-Kram D, Mccullough C, et al. Use of Transgenic Animals for Carcinogenicity Testing:Considerations and Implication for Risk Assessment[J]. Toxicol Pathol, 2000, 28: 482-499. DOI:10.1177/019262330002800320 |
| [39] |
D Vries A, Van Oostrom C, Dortant P, et al. Spontaneous Liver Tumors and Benzo[a] Pyrene-induced Lymphomas in XPA-deficient Mice[J]. Mol Carcinog, 1997, 19: 46-53. DOI:10.1002/(SICI)1098-2744(199705)19:1<46::AID-MC7>3.0.CO;2-L |
| [40] |
Boverhof D, Chamberlain M, Elcombe C, et al. Transgenic Animal Models in Toxicology Historical Perspectives and Future Outlook[J]. Toxicol Sci, 2011, 121: 207-233. DOI:10.1093/toxsci/kfr075 |
| [41] |
Storer R, Sistare F, Reddy M, et al. An Industry Perspective on the Utility of Short-term Carcinogenicity Testing in Transgenic Mice in Pharmaceutical Development[J]. Toxicol Pathol, 2009, 38: 51-61. |
| [42] |
Paranjpe M, Belich J, Mann P, et al. A Comparison of Spontaneous Tumors in Tg.rasH2 Mice in 26-week Carcinogenicity Studies Conducted at a Single Test Facility During 2004 to 2012 and 2013 to 2018[J]. Toxicol Pathol, 2019, 47(1): 18-25. DOI:10.1177/0192623318810202 |
| [43] |
宋征, 徐景宏, 王庆利, 等. 转基因小鼠在药物致癌性评价中的应用[J]. 中国药理学与毒理学杂志, 2010, 24(6): 557-561. |
| [44] |
FDA. Pharmacology/Toxicology Review: Plecanatide[R]. NDA 208745. CDER, 2017.
|
| [45] |
FDA. Pharmacology/Toxicology Review: Baricitinib[R]. NDA 207924. CDER, 2018.
|
| [46] |
Prashant R, Namblar, and Daniel M, et al. Regulatory Forum Commentary Counterpoint:Dose Selection for RasH2 Mouse Carcinogenicity Studies[J]. Toxicol Pathol, 2015, 43(5): 628-632. DOI:10.1177/0192623315578012 |
| [47] |
Jarig Darbes, Frank D Sistare, Joseph J D. Regulatory Forum Commentary Counterpoint:Dose Selection for RasH2 Mouse Carcinogenicity Studies[J]. Toxicol Pathol, 2015, 43(5): 621-627. DOI:10.1177/0192623315587722 |