 2019, Vol. 33
2019, Vol. 33 

头孢克肟是日本藤泽药品工业株式会社研制开发的第三代头孢产品, 1987年在日本上市, 我国1994年开始进口原料及制剂, 国产制剂于2001年上市。现行《中华人民共和国药典》(以下简称《中国药典》)2015年版二部中收录了头孢克肟胶囊、头孢克肟颗粒和头孢克肟片三种口服固体制剂[1]。
作为第三代头孢菌素, 头孢克肟的抗菌谱包括链球菌、肺炎链球菌、淋球菌、大肠杆菌、克雷白杆菌、卡他布拉汉菌、沙雷杆菌、枸橼酸杆菌、阴沟肠杆菌、产气杆菌、流感嗜血杆菌等。作用机制为阻止细菌细胞壁的合成, 与青霉素结合蛋白(PBP)中PBP1和PBP3有较高亲和性, 对细菌产生的β-内酰胺酶稳定。常用于敏感菌引起的呼吸道感染、胆道感染、尿道感染、猩红热、中耳炎、鼻旁窦(副鼻窦)炎。其对金黄色葡萄球菌ATCC25923的质控范围为8~32 μg·mL-1, 对大肠埃希菌ATCC25922为0.25~1 μg·mL-1。根据药品说明书, 头孢克肟片剂200 mg单剂口服, 血浆峰浓度为3.7 μg·mL-1, 达峰时间为2~6小时, 血清蛋白结合率为65%, 24小时内吸收药物的50%以原形从尿中排出, 连续服药14天, 无体内蓄积作用, 不受饮食影响。生物药剂学特征属于低溶解性、低渗透性药物, 生物药剂学分类为Ⅳ类(BCS Class Ⅳ)。
本研究拟通过计算机模拟与预测技术, 结合头孢克肟在小鼠体内的生理药代动力学文献数据[2-3], 对头孢克肟口服固体制剂的生物药剂学及药理特征进行整体研究:以静脉注射途径对品种在体内的药代动力学参数进行研究, 通过口服途径摸索其生物药剂学特征, 建立头孢克肟口服制剂-在不同种属体内的生理药代动力学(Physiologically Based Pharmacokinetic, PBPK)模型, 计算头孢克肟不同口服制剂在体内的药代动力学过程, 结合PBPK模型对头孢克肟在人体内的释放过程进行描述与计算, 以体外溶出曲线为影响因子, 评价头孢克肟固体口服制剂体外溶出度与体内有效性的相关关系。
1 材料与方法 1.1 仪器与试剂FODT-601光纤药物溶出度实时测定仪; 液质联用系统:DIONEX UltiMate 3000, Agilent C8色谱柱(4.6 mm×150 mm, 5 μm); GastroPlus软件(9.0.0007, 美国Simulations公司), 包含有Basic、PBPK、IVIVC、PKPlus、Metabolism & Transport、ADMET Predictor和Optimization Biologics Module模块。
1.2 实验动物普通级比格犬, 8~1 2月龄, 体重范围为7.78~9.48 kg, 饲育条件:连体不锈钢犬笼(900 mm×1000 mm×2080 mm), 每12小时交替光照, 控制温度16~26℃, 相对湿度40%~70%, 换气次数10~20次/小时。
1.3 标准物质信息头孢克肟对照品(批号130503-201205, 含量89.2%)、替硝唑对照品(批号100336-200703, 含量99.8%)均为中国食品药品检定研究院提供。
1.4 供试品信息头孢克肟胶囊(规格100 mg, 批号AG031);头孢克肟片(规格100 mg, 批号2170001);头孢克肟颗粒(规格50 mg, 批号G1707001);头孢克肟细粒剂(规格100 mg, 批号AJ231)。
1.5 LC-MS试验参数流动相:0.5%甲酸水溶液-0.1%甲酸乙醇溶液(65:35), 流速1.0 mL·min-1, 柱温40℃, 进样体积2 μL; 质谱方式为电喷雾离子化源(ESI), 正离子检测, SV:3500, VT:500, SG:75, AG:20, CT:400, T-lens:107/89, CE:16, 扫描模式为反应监测(SRM)。
样品处理方法:精密量取血浆样品50 μL, 加入浓度为4000 ng·mL-1的替硝唑(内标)甲醇溶液50 μL, 加入甲醇300 μL, 混合均匀后于每分钟12000 rpm离心10 min, 精密量取上清液100 μL, 测定。
血药浓度质谱测定时间点:口服给药后第0、0.5、1、2、2.5、3、3.5、4、4.5、5、5.5、6、7、8、10、12、24小时进行采血样本的测定。
2 结果与讨论 2.1 头孢克肟血药浓度测定质谱方法学结果利用质谱方法对血液中的头孢克肟(母离子454.064, 子离子284.928)进行定量, 以替硝唑(母离子248.060, 子离子121.138)为内标物, 进行方法学考察。结果显示在浓度20~20000 ng·mL-1, 样品与内标峰面积比与浓度具有良好线性关系, R=0.9998;精密度中相对误差(RE)为-6.02%, 批内精密度(RSD)为6.22%, 批间精密度(RSD)为7.19%;线性范围内头孢克肟绝对回收率为96.30%, RSD为1.56%, 内标回收率为97.20%, RSD为2.37%。均符合相关生物样品分析方法确证的要求。
2.2 头孢克肟PBPK模型的建立利用GastroPlus软件ADMET模块, 计算头孢克肟生物药剂学参数, 建立头孢克肟PBPK基本参数模型; 以其为依据创建头孢克肟在小鼠、比格犬体内静脉注射给药的生理药代动力学模型, 以文献数据对模型中涉及的头孢克肟生物药剂学参数进行验证(表 1)[2-3], 小鼠和比格犬静脉给药PBPK模型计算结果与文献数据间的回归系数R2分别为0.83和0.80(图 1)。
|
|
表 1 头孢克肟生物药剂学参数 |


|
图 1 头孢克肟在小鼠、比格犬体内静脉给药的PBPK模型验证结果 |
以比格犬为研究对象, 以LC-MS为检测方法, 分别对头孢克肟片、头孢克肟胶囊、头孢克肟细粒剂和头孢克肟颗粒剂口服给药100 mg的24小时血药浓度进行测定(表 2)。以头孢克肟在比格犬体内的PBPK模型为基础, 分别建立上述4种制剂的数据模型, 通过比格犬口服头孢克肟制剂后血药浓度数据对该品种不同制剂在体内的吸收、分布、代谢、排泄特征进行确证。
|
|
表 2 头孢克肟口服制剂在比格犬体内的血药浓度结果 |
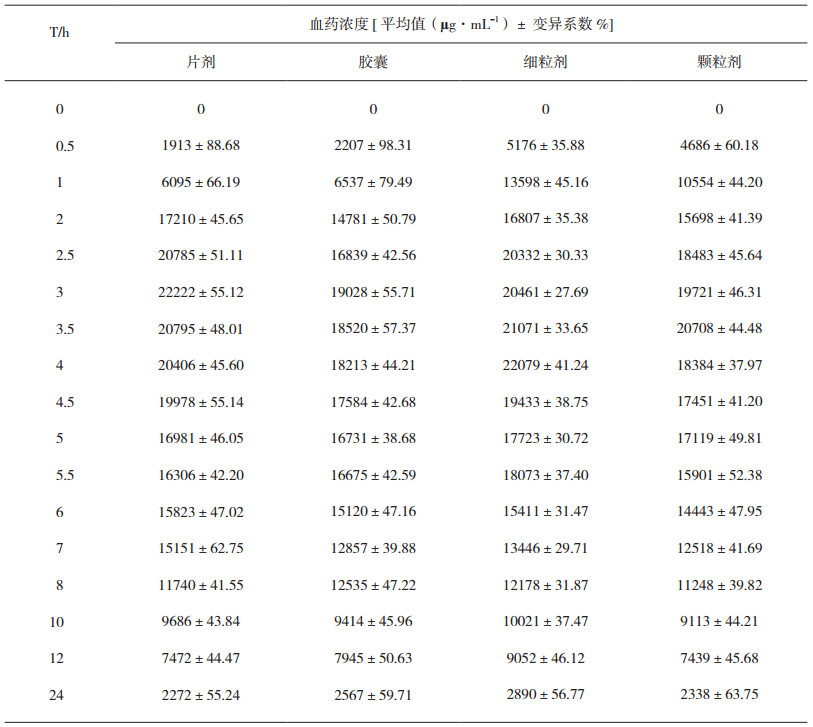
将上述不同剂型的头孢克肟口服制剂在比格犬体内的血药浓度数据加载至头孢克肟-比格犬生理药代动力学模型, 配合对应口服制剂特征参数, 进行模型计算, 结果头孢克肟-比格犬生理药代动力学模型对应片剂、胶囊剂、细粒剂和颗粒剂的模型准确性以计算药时曲线与实测结果间回归系数R2计分别为0.909、0.907、0.920和0.964(图 2)。

|
图 2 头孢克肟口服制剂在比格犬体内数据的PBPK模型验证结果 |
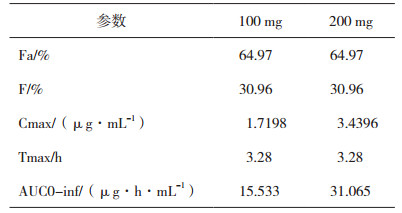
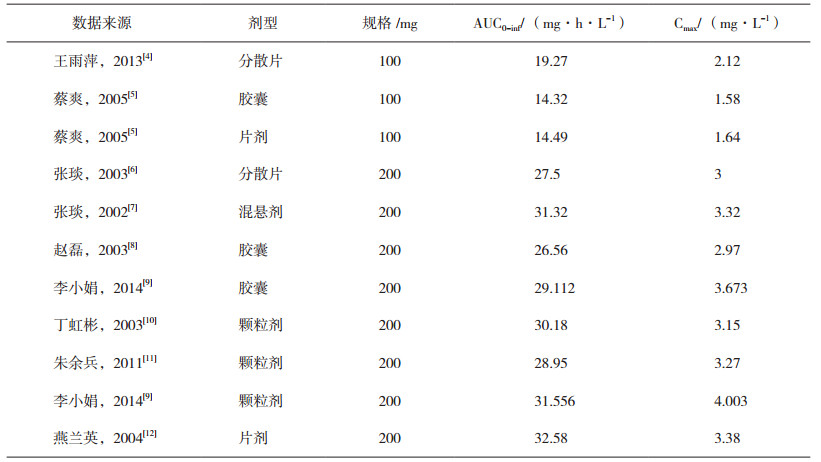
以比格犬生理药代动力学模型为基础, 利用GastroPlusTM软件中的种属外推经验公式, 建立头孢克肟口服制剂在人体内的PBPK模型。利用该模型分别计算人口服100 mg规格、200 mg规格制剂对应药代动力学参数, 其中Fa为口服吸收率、F为口服绝对生物利用度(表 3), 拟合相应规格口服制剂在人体内的药时曲线(图 3)。现有文献报道的规格为100 mg、200 mg头孢克肟口服制剂药代动力学试验结果(表 4), 制剂类型包括分散片(Dispersible Tablet, DT)、胶囊(Capsule, C)、片剂(Tablet, T)、混悬剂(Suspension, S)和颗粒剂(Granule, G)。以模型计算的药代动力学参数AUC0-inf和Cmax结果为100%标准, 对上述文献中相应参数结果进行比例分布研究, 9篇文献中11组头孢克肟口服制剂的药代动力学数据均值均分布于模型计算结果的80%至125%范围区间内(图 4)。验证了人口服头孢克肟PBPK模型对于普通口服制剂在相应规格间的准确性与通用性, 可用于普通口服制剂相应规格的体内药代动力学研究。由于PBPK模型参数选用的是亚洲人种(日本)的生理信息, 因此, 选用体内数据均采用亚洲人体内研究结果。
|
|
表 3 不同规格口服制剂对应药代动力学参数结果 |

|
图 3 利用PBPK模型计算不同规格头孢克肟口服制剂的药时曲线 |
|
|
表 4 头孢克肟口服制剂的药代动力学参数结果 |

|
图 4 头孢克肟口服制剂实测AUC和Cmax均值与模型计算结果比较 |
头孢克肟属于低溶解、低渗透的BCS Ⅳ类药物。根据与机体相关的生理学性质、药物理化性质和PBPK模型结果, 通过机制性卷积分和去卷积分方法, 模拟头孢克肟口服制剂的药代动力学和药效动力学行为, 利用GastroPlusTM软件模块计算在不同器官部位的吸收分布情况(图 5), 结果显示人口服头孢克肟的主要吸收部位为空肠(Jejunum)。根据头孢克肟口服制剂在人体内的药代动力学数据, 计算累计吸收曲线, 分别以门静脉吸收量、体循环吸收量和累计吸收量进行统计(图 6)。

|
图 5 人口服头孢克肟的体内吸收分布图 |

|
图 6 人口服头孢克肟的体内累计吸收量曲线 |
头孢克肟口服制剂在《中国药典》2015年版、日本药典17版[13]和美国药典41版[14]中均有收载, 其中我国药典收录了片剂、胶囊和颗粒剂三种剂型, 用于溶出度检查的介质均为pH 7.2缓冲液, 转速分别为每分钟100转、每分钟100转和每分钟50转, 取样时间均为30分钟, 溶出限度均为80%;日本药典收载了胶囊剂的溶出度方法, 对应条件为pH 7.5缓冲液, 每分钟50转, 90分钟取样和80%限度; 美国药典则只对片剂进行溶出度要求, 条件为pH 7.2缓冲液, 每分钟100转, 45分钟取样和75%限度。将人口服制剂PBPK模型计算的累计吸收曲线与上述各药典标准限度进行比较(图 7), 各国药典溶出度检查法规定在相应取样时间与对应限度均满足头孢克肟在人体内累计吸收量的要求。头孢克肟属于生物药剂学分类BCS的Ⅳ类药物, 具有低溶解度和渗透性特征, 通过溶出限度与累计吸收量的比较, 证实满足各国药典质控标准的头孢克肟口服固体制剂, 其渗透性是限制体内吸收的瓶颈环节, 而不受限于相应溶出介质条件下的溶出量。

|
图 7 溶出限度与累计溶出、吸收量比较 |
日本橙皮书收载了头孢克肟细粒剂和胶囊的参比溶出曲线, 其中胶囊剂为50 mg规格, 根据橙皮书, 头孢克肟在pH 1.2至pH 7.5的溶出介质中, 溶出度逐渐增加, 溶解度随缓冲液系统的pH值增加而增加。本项研究中, 以规格为100 mg的头孢克肟胶囊为供试品, 参照日本橙皮书中溶出系统, 测定了pH 1.2、pH 6.8、pH 7.5和水为溶出介质条件下的溶出曲线(图 8), 结果与橙皮书中50 mg规格产品的溶出曲线类似。根据人口服头孢克肟PBPK模型中生理参数信息, 在空腹口服条件下, 头孢克肟制剂在胃中滞留时间约为0.25小时, 对应器官内条件为pH 1.2, 之后从十二指肠至结肠的器官内条件为pH值均大于6.0。根据溶出度结果, 头孢克肟在pH 1.2介质条件下4小时溶出量达到60%, 在pH 6.8介质条件下3小时溶出量接近100%(图 9)。通过比较, 制剂在pH 1.2环境下0.25小时溶出量满足累计吸收量的要求, 而pH 6.8条件下的溶出曲线反映制剂在肠道过程中的溶出量满足累计吸收量的要求。结果表明该批头孢克肟胶囊100 mg规格能够满足体内累计吸收量的要求, 可以满足制剂在体内各器官累计吸收的要求。

|
图 8 头孢克肟胶囊(100 mg)的实测溶出曲线 |

|
图 9 人口服头孢克肟PBPK模型生理参数与溶出曲线比较图 |
头孢克肟在人体内的转运过程可能受P-糖蛋白(P-glycoprotein, P-gp)影响, 在上述模型建立和验证过程中假设了制剂辅料中不包含对P-糖蛋白可能造成影响的因素及忽略了药物与肠道肽转运蛋白(PEPT1)转运体的特异转运作用[15]。
3 结论在仿制药评价研究中, 溶出曲线相似性比较是口服固体制剂有效性研究的主要手段。将相同剂型、相同规格的参比制剂与仿制制剂在不同溶出介质条件下的溶出曲线进行相关关系计算, 得到两种制剂溶出行为的相似程度结果, 以此作为仿制制剂有效性评判的标准[16-17]。在本研究中, 将同品种不同口服固体制剂作为一个整体研究, 对头孢克肟品种的结构、理化性质、药代动力学特征和生物药剂学参数进行研究和确证, 通过静脉注射、口服等不同制剂给药途径, 以比格犬和人体分别作为受试对象, 以实测体内数据与文献数据验证相结合, 建立并验证头孢克肟在人体内的生理药代动力学模型, 进一步延伸至不同剂型、不同规格的体内模型并通过数据验证。以上述模型为基础, 模拟口服头孢克肟在人体内的吸收、分布、代谢、消除过程, 计算品种口服的药时曲线, 拟合累计吸收量的时间曲线, 并以此作为口服制剂有效性评价的通用指标。相较单纯的溶出曲线比较方法, 本研究建立的有效性评价方法能够建立溶出度行为与药物体内吸收的关系, 具有更好的体内外相关性, 提高体外有效性评价的可靠性和准确度, 同时大幅度降低仿制药评价的过程成本, 提高整体研究效率。
| [1] |
中华人民共和国药典: 第二卷[S].北京: 中国医药科技出版社, 2015: 257.
|
| [2] |
H Sakamoto, T Hirose, S Nakamoto, et al. Pharmacokinetics of FK482 Anew Orally Active Cephalosporin in Animals[J]. The Journal of Antibiotics, 1988, 12: 1896-1905. |
| [3] |
Meir Bialer, Vijay K Batra, John A Morrison, et al. Dosedependent Pharmacokinetics of a New Oral Cephalosporin Cefixime in the Dog[J]. Pharmaceutical Research, 1987, 4(1): 33-37. DOI:10.1023/A:1016473709720 |
| [4] |
王雨萍, 鲁卓林, 尹永强, 等. 人血浆中头孢克肟的LC-MS/MS法测定及生物等效性研究[J]. 天津药学, 2013, 25(2): 1-3, 78. DOI:10.3969/j.issn.1006-5687.2013.02.001 |
| [5] |
蔡爽, 冯婉玉, 林建阳, 等. 头孢克肟片和胶囊在健康人体的生物等效性[J]. 中国临床药理学杂志, 2005, 21(5): 375-377. |
| [6] |
张琰, 贺建荣, 文爱东, 等. 头孢克肟分散片在健康志愿者体内生物等效性[J]. 中国医院药学杂志, 2003, 23(7): 388-390. DOI:10.3321/j.issn:1001-5213.2003.07.002 |
| [7] |
张琰, 程建峰, 文爱东, 等. 头孢克肟混悬剂与颗粒剂的药物动力学及生物等效性[J]. 中国抗生素杂志, 2002, 27(11): 681-684. DOI:10.3969/j.issn.1001-8689.2002.11.012 |
| [8] |
赵磊, 文爱东, 张三奇, 等. 头孢克肟胶囊在健康人体内的相对生物等效性[J]. 第四军医大学学报, 2003, 24(7): 661-664. DOI:10.3321/j.issn:1000-2790.2003.07.027 |
| [9] |
李小娟, 李杰, 舒成仁. 头孢克肟颗粒和胶囊在健康人体的生物等效性[J]. 中国药师, 2014, 17(1): 19-22. |
| [10] |
丁虹彬, 张静, 王蓓, 等. 头孢克肟颗粒在健康受试者体内的生物利用度及其药代动力学[J]. 江苏药学与临床研究, 2003, 11(6): 11-14. DOI:10.3969/j.issn.1673-7806.2003.06.005 |
| [11] |
朱余兵, 任军, 于翠霞, 等. 头孢克肟颗粒在健康受试者体内血浓度测定及相对生物利用度研究[J]. 中国药师, 2011, 14(12): 1727-1730. DOI:10.3969/j.issn.1008-049X.2011.12.007 |
| [12] |
燕兰英, 张静, 刘云, 等. 头孢克肟片在健康受试者体内的生物利用度和生物等效性研究[J]. 抗感染药学, 2004, 1(3): 120-122. |
| [13] |
The United States Pharmacopieial Convention. The United States Pharmacopeia: General Chapters[S]. 41st Edition. Rockvill: The United States Pharmacopieial Convention, 2018: 3267.
|
| [14] |
The Ministry of Health, Labour and Welfare. The Japanese Pharmacopoeia[S]. Seventeenth Edition. 2016: 624-625.
|
| [15] |
刘志浩, 刘克辛. 肠道药物转运体及其研究方法[J]. 药学学报, 2011, 46(4): 370-376. |
| [16] |
国家食品药品监督管理总局.普通口服固体制剂参比制剂选择和确定指导原则[EB/OL].[2019-07-01]. http://www.sda.gov.cn/WS01/CL0087/147583.html.
|
| [17] |
国家食品药品监督管理总局.普通口服固体制剂溶出度试验技术指导原则[EB/OL].[2019-07-01]. http://www.sda.gov.cn/WS01/CL1757/114288.html.
|