 2018, Vol. 32
2018, Vol. 32 
复方盐酸替利定口服液属阿片类镇痛药,主要由盐酸替利定(40 mg·mL-1,简称盐酸反式替利定,其化学结构见图 1)和盐酸纳洛酮(5 mg·mL-1)组成。其活性成分替利定口服吸收迅速完全,但它本身不具有镇痛作用,在肝脏中代谢为去甲替利定(Nortilidine,其化学结构见图 1)而产生具有吗啡样的镇痛活性[1-2],其镇痛强度与哌替啶相当, 约为吗啡的1 /10[3-5]。

|
图 1 替利定和去甲替利定的化学结构 |
复方替利定口服液为Warner-Lambert公司开发的阿片类镇痛药,常用于手术、烧伤、癌症以及其他原因引起的中度、重度疼痛的镇痛。该药于1970年首次在德国上市后因其镇痛效果好,口服吸收迅速完全,不良反应较轻等优势在欧洲国家得到了广泛认可[6],已在多个国家注册。近年来,替利定的世界临床医疗用量的增加趋势远远超过了其他各种阿片类镇痛药[7-8],目前复方盐酸替利定口服液尚未在中国上市,国内仍处于申报状态。
笔者在对复方盐酸替利定口服液进行质量研究时,发现有3个杂质的含量较大。本文通过LCMS、IR、H-NMR推测其结构,采用分离、合成等方法获得了这3个杂质,最终确定这3个杂质均由盐酸替利定原料引入。杂质1为盐酸替利定异构体—顺式替利定,杂质2为活性代谢物反式去甲基替利定,杂质3为顺式去甲基替利定(化学结构见图 2)。上述所确定的3个主要杂质的结构与最近欧洲药典(EP5)公布的盐酸替利定的主要杂质结构一致。由于本口服液的质量标准未收入国内外药典,其质量控制方法也未见任何相关文献报道,揭示了杂质的来源之后,为进一步减少杂质的含量,笔者在替利定原料的制备工艺上做了相应改进,一定程度上提高了复方盐酸替利定口服液的质量。

|
图 2 复方替利定口服液中3种杂质的化学结构 |
岛津Shimadzu LC-10A高效液相色谱仪;Agilent 1100LC/MSD液质联用色谱仪;Varian 8 0 M,Avance 300 M核磁共振谱仪[氘带氯仿(CDCl3)为溶剂,四甲基硅烷(TMS)为内标];美国PE公司Spectrum One PE983型红外光谱仪。紫外可见分光光度计:岛津UV-160紫外-可见分光光度仪。
复方盐酸替利定口服液(40 mg·mL-1)(四川抗菌素工业研究所);盐酸替利定原料(自制批号:20130324);盐酸纳洛酮原料(北京四环医药)。
2 复方盐酸替利定口服液及其原料的杂质考察 2.1 高效液相色谱条件岛津LC-10A高效液相色谱仪;Phenomenex Luna辛烷基键合硅胶色谱柱(4.6 mm ×250 mm,5 μm);甲醇-0.2%碳酸氢铵水溶液(70:30,v/v)为流动相;流速1 mL·min-1;检测波长229 nm;柱温35 ℃;进样量20μL[9-10]。
2.2 复方盐酸替利定口服液和盐酸替利定原料杂质高效液相色谱对比结果按照“2.1”节的色谱条件,分别用本品以及与之相应的盐酸替利定和盐酸纳洛酮原料进行色谱对照试验,结果表明,复方盐酸替利定口服液中盐酸替利定的保留时间在约14.6 min,盐酸纳洛酮的保留时间在约5.5 min;最大单个杂质的保留时间10.8 min(简称杂3),其归一化含量约0.2%,其它任何单个杂质的归一化含量均低于0.2%。通过液相色谱比较,复方替利定口服液和盐酸替利定原料3个含量较大的杂质保留时间均一致(图 3~5)。

|
图 3 盐酸替利定原料HPLC图 |

|
图 4 盐酸纳洛酮HPLC图 |

|
图 5 复方替利定口服液HPLC |
为了进一步确证杂质结构,便于做液-质联用分析,将本品用0.1 mol·L-1氢氧化钠溶液破坏后作为供试品(该样品中杂质1、2的量明显增大)。量取该破坏样品适量,用流动相稀释为含盐酸替利定约1 mg·mL-1和盐酸纳洛酮约0.08 mg·mL-1的溶液作为供试液,对破坏后样品进行杂质量的考察。
在“2.1”节色谱条件下采用液-质联用仪(型号为Agilent 1100LC/MSD)对本品进行杂质测定(结果见图 6~9)。

|
图 6 复方盐酸替利定口服液液-质联用液相图谱 |

|
图 7 液质联用杂质1质谱图(保留时间26.7min) |

|
图 8 液质联用杂质2质谱图(保留时间7.3min) |

|
图 9 液质联用杂质3质谱图(保留时间11.5min) |
液质联用试验结果表明,保留时间为26.7min(杂质1)的M+1峰为274.2,因而该杂质的分子量应为273.2,与替利定的分子量相同,可初步推断为顺式替利定。保留时间为7.3 min(杂质2)、11.5 min(杂质3)的两杂质的M+1峰均为260.2,因而两杂质的分子量均应为259.2。鉴于替利定与顺式替利定的叔氮上易于脱去甲基,脱甲基后的分子量均为259.2,因此,可初步推测两杂质可能互为去甲基替利定的顺、反异构体。
2.4 杂质1、2、3的分离及结构鉴定 2.4.1 杂质1的分离及结构鉴定取顺反替利定混合物(含顺式替利定约40%)5.0 g,以二氯甲烷-甲醇(90:1,v/v)作洗脱剂进行柱层析,获得顺式替利定1.2 g。与杂质1的高效液相色谱保留时间一致,并通过红外、质谱、核磁共振分析杂质1为顺式替利定(红外图谱见图 10)。

|
图 10 杂质1紫外图谱 |
解析与结论:杂质1在254 nm左右产生苯环的B带特征吸收,从而表明本品中含有苯环,由π→π*电子跃迁引起(供试液浓度为0. 5 mg·mL-1)。
MS(ESI)274.2(M+1)
碎片(m/e):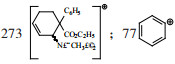
IR(KCl)cm -1:2979(-CH3)强度中等,1723(-C=O)强度强, 1233(-C-O-C-)强度强,694(苯环)强度弱。
1H-NMR(300MHz,CDCl3):δ1.22(3H,t), δ1.91(2H,m), δ2.54(2H,t), δ2.84(3H,s), δ3.02(3H,s), δ4.28(2H,m), δ4.62(1H,d), δ6.05(1H,m), δ6.14(1H,m), δ7.29(1H,m), δ7.33(2H,m), δ7.37(2H,m)。
2.4.2 杂质2、3的合成及结构鉴定取5.4 g反式替利定溶于50 mL氯仿中,冷却到-10℃。搅拌下缓慢将溴水滴入氯仿溶液,滴完后-10℃下反应30 min。将橘红色的反应液倒入4倍体积的蒸馏水中,室温下搅拌,直到反应液变为无色。用氨水将产物pH值调节到10~12,有机层水洗两次,无水硫酸镁干燥。过滤,蒸干溶剂,得到5.2 g固体。-10 ℃滴加7.9 mol·L-1氯化氢乙醇溶液(28 mL),滴毕冰浴搅拌2 h后减压蒸除溶剂,剩余物经THF重结晶,得白色结晶性粉末反式去甲替利定3.7 g[11](顺式去甲替利定制备方法同反式去甲替利定,所用原料为顺式替利定,其他条件不变,制备工艺如图 11)。

|
图 11 杂质2、3的合成路线 |
杂质2:MS(ESI)260.2(M+1)
IR(KCl)cm-1:3433(-NH)强度中等,2970(-CH3)强度强,1724(-C=O)强度强,1181(-C-O-C-)强度强,696(苯环)强度中等。
1H-NMR(300MHz,CDCl3):δ1.1 5(3H,t), δ2.13(t,1H),2.49(2H,t),δ2.11(3H,m),δ2.95(m,2H),2.58(2H,t),δ4.13(2H,m),δ4.22(1H,d),δ6.03(1H,m),δ6.16(1H,m),δ7.36(1H,m),δ7.43(2H,d),δ7.50(2H,m),δ9.51(1H,s)。
杂质3:MS(ESI)260.2(M+1)
IR(KCl)cm-1: 3423(-NH,)强度中等,2965(-CH3)强度强,1722(-C=O)强度强, 1260(-C-O-C-)强度强,697(苯环)强度中等。
1H-NMR(300MHz,CDCl3):δ1.22(3H,t),δ1.96(m,1H),2.42(2H,t),δ2.34(3H,m),δ2.50,2.61(2H,t),δ4.36(2H,m),δ4.41(1H,d),δ6.03(1H,m),δ6.14(1H,m),δ7.29(1H,m),δ7.33(2H,d),δ7.36(2H,m),δ10.42(1H,s)。
杂质2、3氢谱解析与结论:顺势去甲替利定处在同一平面的氢(7,9,10)由于其空间较为拥挤以及受苯环的去屏蔽影响,其化学位移比相应位的反式去甲基替利定的氢向低场移动,另一平面上的氢(12,13,15,16)则向高场移动;而质子个数及其它位的化学位移则基本一致。从而表明杂质2为反式去甲基替利定,杂质3为顺式去甲基替利定。
3 替利定原料工艺改造通过LC-MS和核磁图谱数据,可以确定复方盐酸替利定口服液中3个含量较大的杂质分别为顺式替利定和反、顺式去甲替利定。杂质1为制备替利定原料时产生而分离时残留的部分顺式替利定[12](盐酸替利定原料制备工艺如图 12),反、顺式去甲替利定为制备替利定原料时的降解产物。由此可以推测通过对盐酸替利定原料进行工艺改进,提高原料的纯度,从而提高复方替利定口服液的质量。

|
图 12 盐酸替利定原料制备工艺[12] |
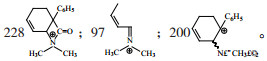
在氮气保护下避光将4(59.5g,0.34 mol)、6(50.5g,0.36 mol)和甲苯(120 mL,除水处理)加入到反应瓶中,加热回流搅拌1 h。-5~0℃缓慢滴加10%盐酸调至pH 2.5 ~ 3.5,分出酸水层,加氨水调至pH 8.5 ~ 9.5,用乙酸乙酯(100 mL×3)萃取,合并有机层,饱和氯化钠溶液洗涤。经无水硫酸钠干燥后过滤,滤液减压蒸除溶剂,得深黄色液体7(80.2g,86.3%)。纯度:反式36.9%,顺式39.7%[12]。
3.2 分离(±)-2-(二甲胺基)-1-苯基-3-环己烯-1-羧酸乙酯(7)工艺改进将7(77.3 g,0.28 mol)溶于无水乙醇(150 mL)中,加入1, 5-萘二磺酸(23.0 g,0.08 mol)的无水乙醇(90 mL)溶液中,-5~0℃搅拌析晶1 h,过滤,得黄色固体65.8 g。加入1 mol·L-1 NaOH溶液462 mL,用乙酸乙酯萃取3次(200 mL×3)。合并有机层,依次用蒸馏水和饱和氯化钠洗涤,无水硫酸钠干燥,过滤,滤液减压蒸馏,得浅黄色液体8(22.8 g,23.5%)[12]。
将8(22.8 g,0.8 mol)溶于200 mL无水乙醇中,-15~-10℃下缓慢滴加氯化氢乙醇溶液(10 mol·L-1,12 mL),减压蒸馏,四氢呋喃重结晶,得白色晶体粉末1(16.8 g,73.7%;杂质1:0.09%;杂质2:0.10%;杂质3:0.18%)[12]。
此步骤用1, 5-萘二磺酸替换1, 5-萘二磺酸二水合物作为拆分剂,可减少反式和顺势替利定的去甲基化,从而减少反、顺式去甲替利定的产生。
4 结论本研究通过对复方替利定口服液杂质的结构确证和替利定原料中杂质的对比,发现其杂质来源于原料的制备过程。在试验过程中,光照有利于顺式替利定的生成,顺反式替利定在有水的环境中更容易去甲基化,所以通过“3.1”、“3.2”节对盐酸替利定原料合成工艺的改进,可以减少顺式替利定的生成和顺反式替利定的去甲基化,从而减少3个杂质的含量,一定程度上提高了复方替利定口服液的质量。此外,1, 5-萘二磺酸比1, 5-萘二磺酸二水合物更易得,价格更低廉,作为拆分剂,在大批量工业化生产时可以有效节约生产成本。
| [1] |
李静, 刘闯, 黄明生. 海洛因依赖者治疗阻抗因素自评量表的编制[J]. 华西医学, 2000, 15: 23-24. DOI:10.3969/j.issn.1002-0179.2000.01.012 |
| [2] |
Sun L, LI Xiao, Liang JC, et al. Randomized and Double-blind Controlled Clinical Trial of Tilidine Hydrochloride Oral Solution for Analgesia[J]. Chinese Journal of Clinical Pharmacology, 2013, 29(1): 6-8. |
| [3] |
陈素青, 黄新洁, 崔艳英, 等. 盐酸替利定的依赖性评价[J]. 中国药物依赖性杂志, 2006, 15(5): 363-366. |
| [4] |
THIERRY C, BOEYNAEMSJM JM, PAOLO M. Actions of Tilidine and Nortilidine on Cloned Opioid Receptors[J]. Eur J Pharmaeol, 2005, 50(3): 205. |
| [5] |
Liu N, MA Tian-Jiang, GY Zhang, et al. Clinical Observation of Oxycodone Hydrochloride Sustained Release Tablets in the Treatment of Moderate and Severe Cancer Pain[J]. China Practical Medicine, 2016(23): 45-49. |
| [6] |
郝光涛, 白少柏, 郑专杰, 等. LC-MS/MS法测定人血浆中替利定和去甲替利定的质量浓度[J]. 药物分析杂志, 2012, 32(10): 1726-1730. |
| [7] |
INCB. Trends in the Licit Movement of Narcaotic Drugs. Narcotic Drugs-Estimated World Requirements of 2001[R]. New York: United Nations, 2001.
|
| [8] |
2016-2022年中国替利定(INN)及其盐行业进口态势分析及对外贸易前景展望报告[R]. 北京: 智研咨询集团, 2016: 5-21.
|
| [9] |
徐明琴, 唐克慧, 蒲刚, 等. RP-HPLC测定复方盐酸替利定口服液含量及有关物质[J]. 中国药学杂志, 2009, 4(20): 1588-1591. |
| [10] |
赵雨润. 复方替利定缓释片的工艺制备与质量控制方法研究[D]. 石家庄: 河北医科大学, 2015. 4-90.
|
| [11] |
S Gerhard, W Watzinger, Herrmann, et al. Dextrorotatory 3r-N-Monomethyl-Amlno-4c-Phenyl-4t-Ethoxycarbonicyclohexenexene-1 and Process for the Production Thereof: US, 38821[P]. (1975-03-06)[2017-04-12].
|
| [12] |
李宏名, 吴林彬, 胡延雷, 等. 盐酸替利定的合成[J]. 中国医药工业杂志, 2012, 43(7): 97-99. |